 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Property Prediction: A Multilevel Quantum Interactions Modeling Perspective
Paper and Code
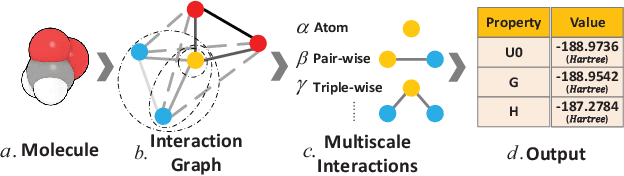
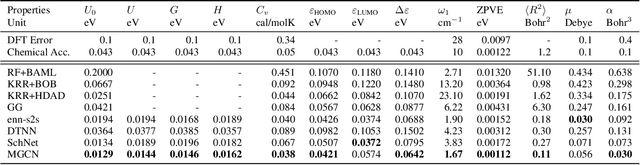
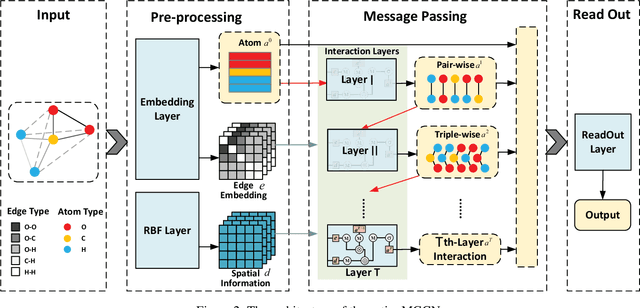
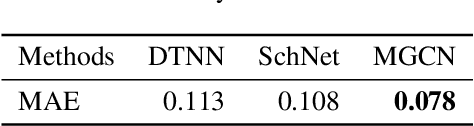
Predicting molecular properties (e.g., atomization energy) is an essential issue in quantum chemistry, which could speed up much research progress, such as drug designing and substance discovery. Traditional studies based on density functional theory (DFT) in physics are proved to be time-consuming for predicting large number of molecules. Recently, the machine learning methods, which consider much rule-based information, have also shown potentials for this issue. However, the complex inherent quantum interactions of molecules are still largely underexplored by existing solutions. In this paper, we propose a generalizable and transferable Multilevel Graph Convolutional neural Network (MGCN) for molecular property prediction. Specifically, we represent each molecule as a graph to preserve its internal structure. Moreover, the well-designed hierarchical graph neural network directly extracts features from the conformation and spatial information followed by the multilevel interactions. As a consequence, the multilevel overall representations can be utilized to make the prediction. Extensive experiments on both datasets of equilibrium and off-equilibrium molecules demonstrate the effectiveness of our model. Furthermore, the detailed results also prove that MGCN is generalizable and transferable for the prediction.