 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTZC: Efficient Inter-Process Communication for Robotics Middleware with Partial Serialization
Mar 01, 2019
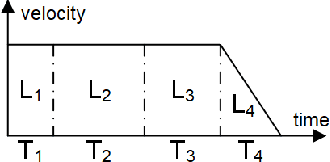
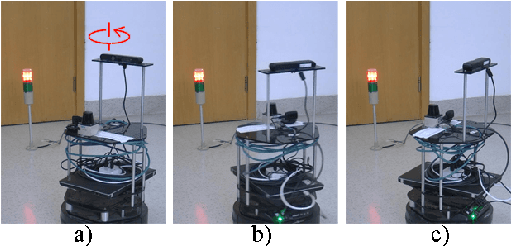
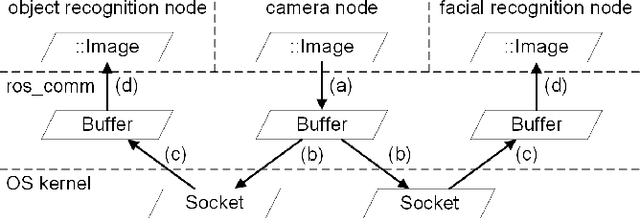
Inter-process communication (IPC) is one of the core functions of modern robotics middleware. We propose an efficient IPC technique called TZC (Towards Zero-Copy). As a core component of TZC, we design a novel algorithm called partial serialization. Our formulation can generate messages that can be divided into two parts. During message transmission, one part is transmitted through a socket and the other part uses shared memory. The part within shared memory is never copied or serialized during its lifetime. We have integrated TZC with ROS and ROS2 and find that TZC can be easily combined with current open-source platforms. By using TZC, the overhead of IPC remains constant when the message size grows. In particular, when the message size is 4MB (less than the size of a full HD image), TZC can reduce the overhead of ROS IPC from tens of milliseconds to hundreds of microseconds and can reduce the overhead of ROS2 IPC from hundreds of milliseconds to less than 1 millisecond. We also demonstrate the benefits of TZC by integrating with TurtleBot2 that are used in autonomous driving scenarios. We show that by using TZC, the braking distance can be shortened by 16% than ROS.
FDR-Corrected Sparse Canonical Correlation Analysis with Applications to Imaging Genomics
Jun 23, 2018
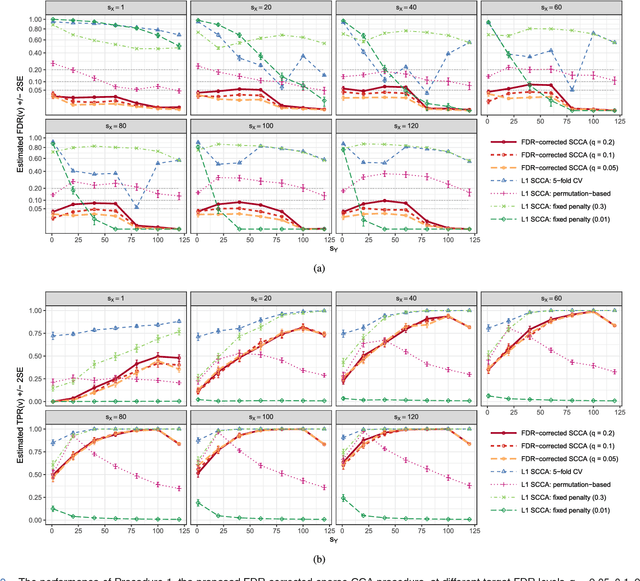
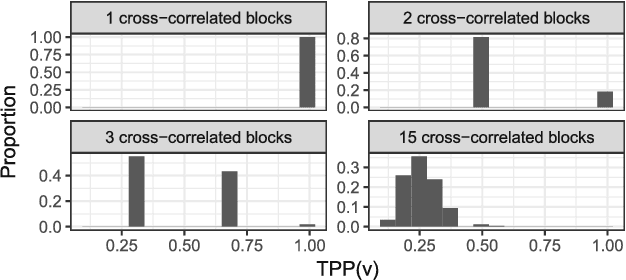
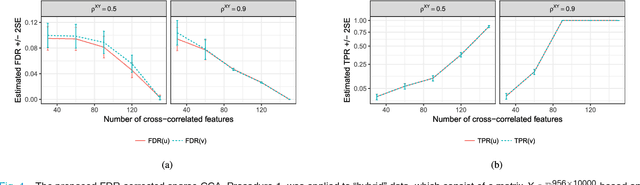
Reducing the number of false discoveries is presently one of the most pressing issues in the life sciences. It is of especially great importance for many applications in neuroimaging and genomics, where datasets are typically high-dimensional, which means that the number of explanatory variables exceeds the sample size. The false discovery rate (FDR) is a criterion that can be employed to address that issue. Thus it has gained great popularity as a tool for testing multiple hypotheses. Canonical correlation analysis (CCA) is a statistical technique that is used to make sense of the cross-correlation of two sets of measurements collected on the same set of samples (e.g., brain imaging and genomic data for the same mental illness patients), and sparse CCA extends the classical method to high-dimensional settings. Here we propose a way of applying the FDR concept to sparse CCA, and a method to control the FDR. The proposed FDR correction directly influences the sparsity of the solution, adapting it to the unknown true sparsity level. Theoretical derivation as well as simulation studies show that our procedure indeed keeps the FDR of the canonical vectors below a user-specified target level. We apply the proposed method to an imaging genomics dataset from the Philadelphia Neurodevelopmental Cohort. Our results link the brain connectivity profiles derived from brain activity during an emotion identification task, as measured by functional magnetic resonance imaging (fMRI), to the corresponding subjects' genomic data.
Kernel Method for Detecting Higher Order Interactions in multi-view Data: An Application to Imaging, Genetics, and Epigenetics
Jul 14, 2017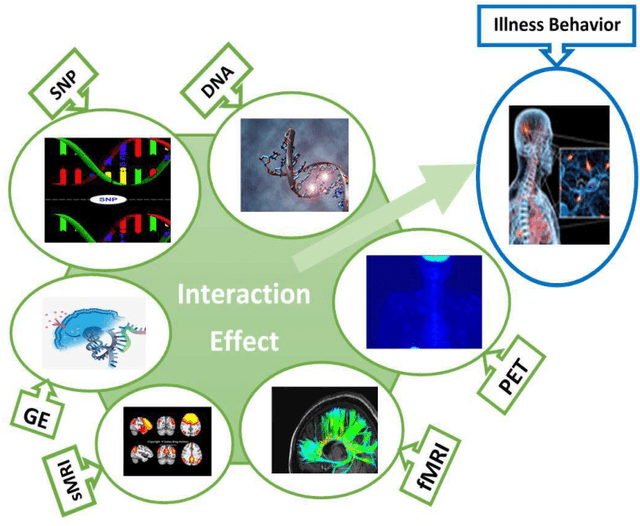
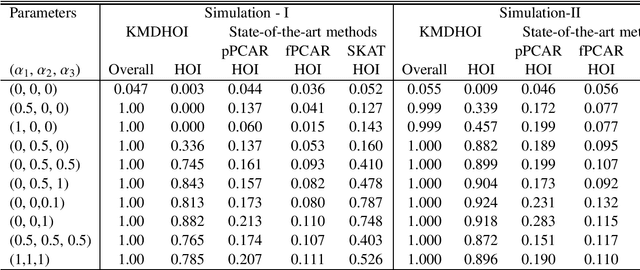
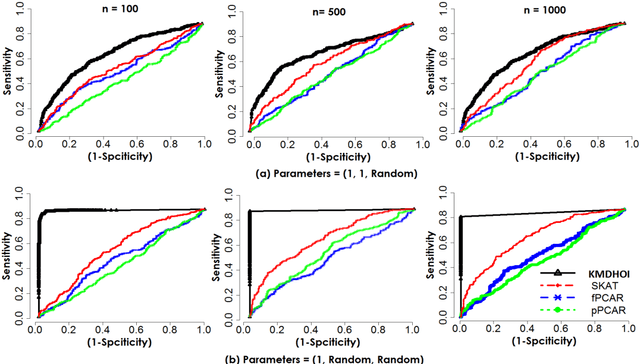
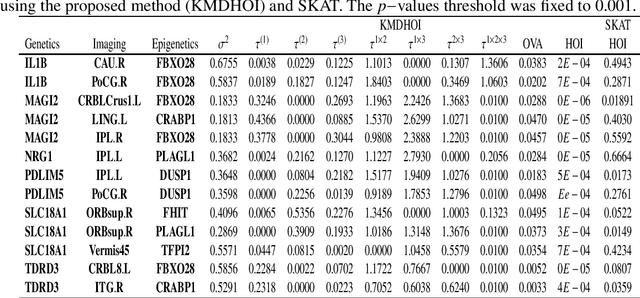
In this study, we tested the interaction effect of multimodal datasets using a novel method called the kernel method for detecting higher order interactions among biologically relevant mulit-view data. Using a semiparametric method on a reproducing kernel Hilbert space (RKHS), we used a standard mixed-effects linear model and derived a score-based variance component statistic that tests for higher order interactions between multi-view data. The proposed method offers an intangible framework for the identification of higher order interaction effects (e.g., three way interaction) between genetics, brain imaging, and epigenetic data. Extensive numerical simulation studies were first conducted to evaluate the performance of this method. Finally, this method was evaluated using data from the Mind Clinical Imaging Consortium (MCIC) including single nucleotide polymorphism (SNP) data, functional magnetic resonance imaging (fMRI) scans, and deoxyribonucleic acid (DNA) methylation data, respectfully, in schizophrenia patients and healthy controls. We treated each gene-derived SNPs, region of interest (ROI) and gene-derived DNA methylation as a single testing unit, which are combined into triplets for evaluation. In addition, cardiovascular disease risk factors such as age, gender, and body mass index were assessed as covariates on hippocampal volume and compared between triplets. Our method identified $13$-triplets ($p$-values $\leq 0.001$) that included $6$ gene-derived SNPs, $10$ ROIs, and $6$ gene-derived DNA methylations that correlated with changes in hippocampal volume, suggesting that these triplets may be important in explaining schizophrenia-related neurodegeneration. With strong evidence ($p$-values $\leq 0.000001$), the triplet ({\bf MAGI2, CRBLCrus1.L, FBXO28}) has the potential to distinguish schizophrenia patients from the healthy control variations.
Influence Function and Robust Variant of Kernel Canonical Correlation Analysis
May 09, 2017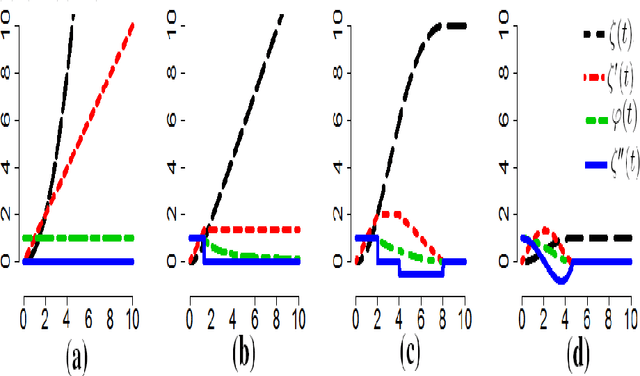
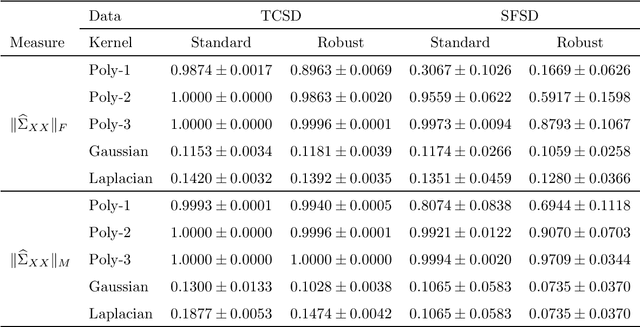

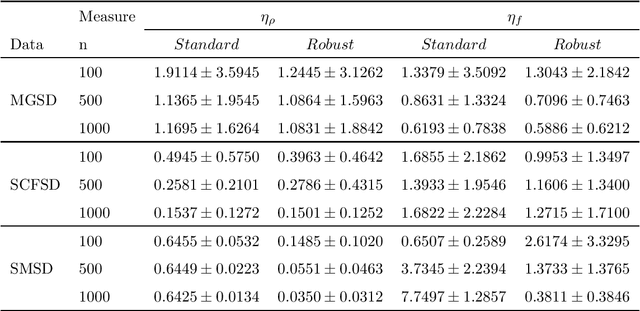
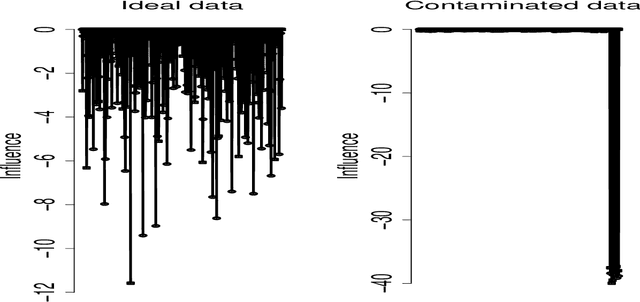
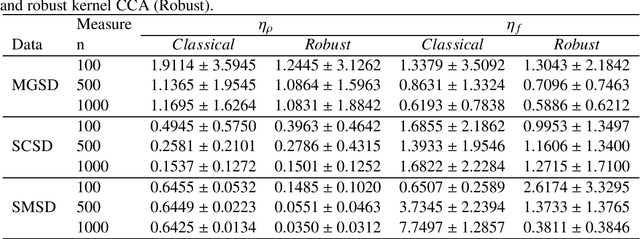
Many unsupervised kernel methods rely on the estimation of the kernel covariance operator (kernel CO) or kernel cross-covariance operator (kernel CCO). Both kernel CO and kernel CCO are sensitive to contaminated data, even when bounded positive definite kernels are used. To the best of our knowledge, there are few well-founded robust kernel methods for statistical unsupervised learning. In addition, while the influence function (IF) of an estimator can characterize its robustness, asymptotic properties and standard error, the IF of a standard kernel canonical correlation analysis (standard kernel CCA) has not been derived yet. To fill this gap, we first propose a robust kernel covariance operator (robust kernel CO) and a robust kernel cross-covariance operator (robust kernel CCO) based on a generalized loss function instead of the quadratic loss function. Second, we derive the IF for robust kernel CCO and standard kernel CCA. Using the IF of the standard kernel CCA, we can detect influential observations from two sets of data. Finally, we propose a method based on the robust kernel CO and the robust kernel CCO, called {\bf robust kernel CCA}, which is less sensitive to noise than the standard kernel CCA. The introduced principles can also be applied to many other kernel methods involving kernel CO or kernel CCO. Our experiments on synthesized data and imaging genetics analysis demonstrate that the proposed IF of standard kernel CCA can identify outliers. It is also seen that the proposed robust kernel CCA method performs better for ideal and contaminated data than the standard kernel CCA.
Learning Schizophrenia Imaging Genetics Data Via Multiple Kernel Canonical Correlation Analysis
Sep 15, 2016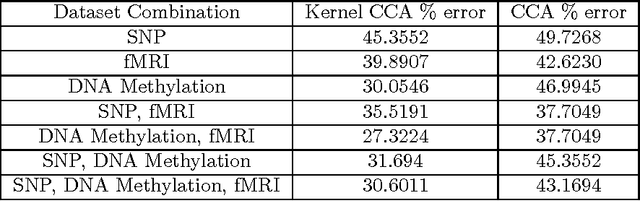
Kernel and Multiple Kernel Canonical Correlation Analysis (CCA) are employed to classify schizophrenic and healthy patients based on their SNPs, DNA Methylation and fMRI data. Kernel and Multiple Kernel CCA are popular methods for finding nonlinear correlations between high-dimensional datasets. Data was gathered from 183 patients, 79 with schizophrenia and 104 healthy controls. Kernel and Multiple Kernel CCA represent new avenues for studying schizophrenia, because, to our knowledge, these methods have not been used on these data before. Classification is performed via k-means clustering on the kernel matrix outputs of the Kernel and Multiple Kernel CCA algorithm. Accuracies of the Kernel and Multiple Kernel CCA classification are compared to that of the regularized linear CCA algorithm classification, and are found to be significantly more accurate. Both algorithms demonstrate maximal accuracies when the combination of DNA methylation and fMRI data are used, and experience lower accuracies when the SNP data are incorporated.
Gene-Gene association for Imaging Genetics Data using Robust Kernel Canonical Correlation Analysis
Jun 01, 2016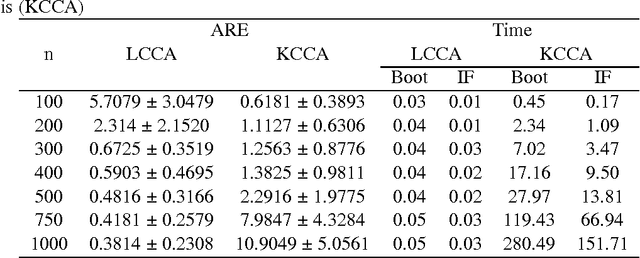
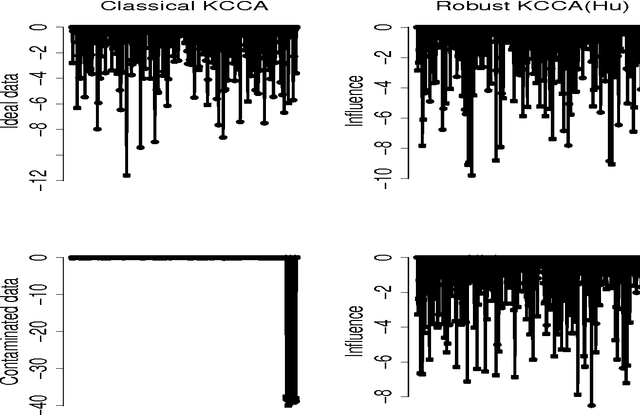
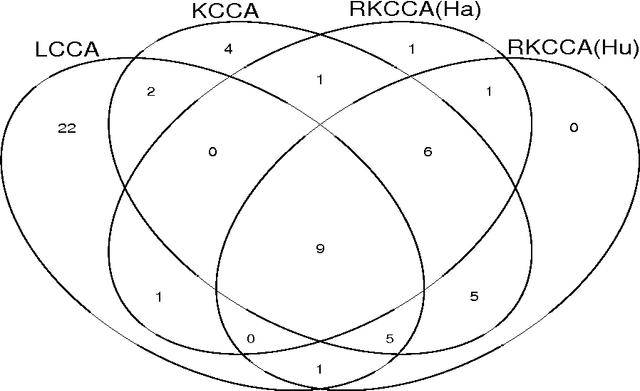
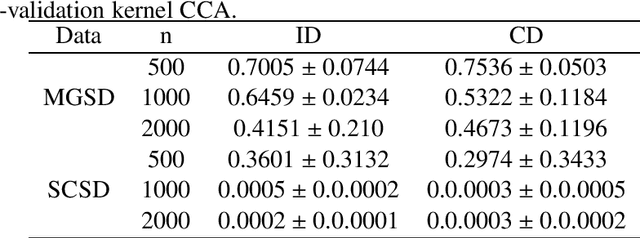
In genome-wide interaction studies, to detect gene-gene interactions, most methods are divided into two folds: single nucleotide polymorphisms (SNP) based and gene-based methods. Basically, the methods based on the gene are more effective than the methods based on a single SNP. Recent years, while the kernel canonical correlation analysis (Classical kernel CCA) based U statistic (KCCU) has proposed to detect the nonlinear relationship between genes. To estimate the variance in KCCU, they have used resampling based methods which are highly computationally intensive. In addition, classical kernel CCA is not robust to contaminated data. We, therefore, first discuss robust kernel mean element, the robust kernel covariance, and cross-covariance operators. Second, we propose a method based on influence function to estimate the variance of the KCCU. Third, we propose a nonparametric robust KCCU method based on robust kernel CCA, which is designed for contaminated data and less sensitive to noise than classical kernel CCA. Finally, we investigate the proposed methods to synthesized data and imaging genetic data set. Based on gene ontology and pathway analysis, the synthesized and genetics analysis demonstrate that the proposed robust method shows the superior performance of the state-of-the-art methods.
Identifying Outliers using Influence Function of Multiple Kernel Canonical Correlation Analysis
Jun 01, 2016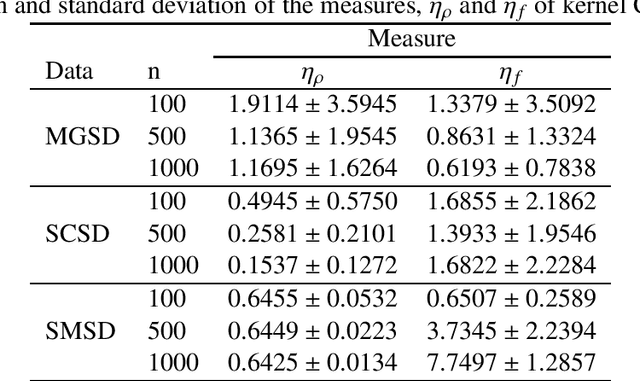
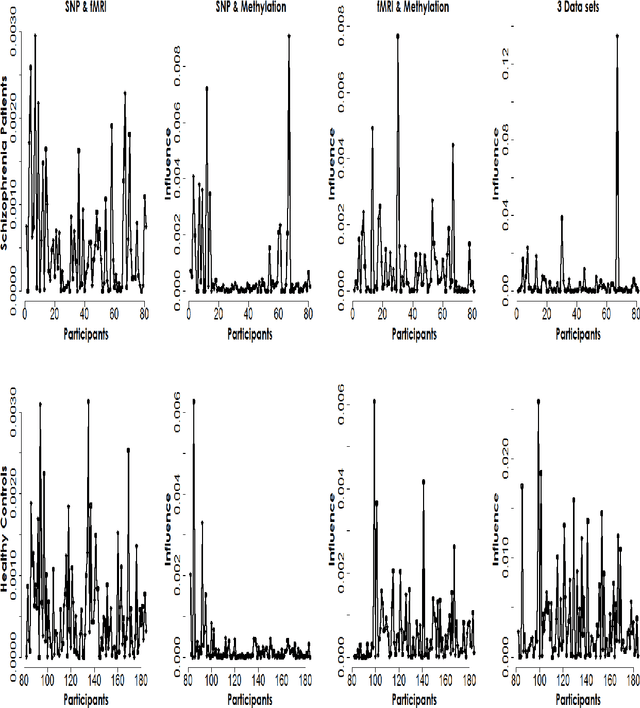
Imaging genetic research has essentially focused on discovering unique and co-association effects, but typically ignoring to identify outliers or atypical objects in genetic as well as non-genetics variables. Identifying significant outliers is an essential and challenging issue for imaging genetics and multiple sources data analysis. Therefore, we need to examine for transcription errors of identified outliers. First, we address the influence function (IF) of kernel mean element, kernel covariance operator, kernel cross-covariance operator, kernel canonical correlation analysis (kernel CCA) and multiple kernel CCA. Second, we propose an IF of multiple kernel CCA, which can be applied for more than two datasets. Third, we propose a visualization method to detect influential observations of multiple sources of data based on the IF of kernel CCA and multiple kernel CCA. Finally, the proposed methods are capable of analyzing outliers of subjects usually found in biomedical applications, in which the number of dimension is large. To examine the outliers, we use the stem-and-leaf display. Experiments on both synthesized and imaging genetics data (e.g., SNP, fMRI, and DNA methylation) demonstrate that the proposed visualization can be applied effectively.
Robust Kernel (Cross-) Covariance Operators in Reproducing Kernel Hilbert Space toward Kernel Methods
Feb 17, 2016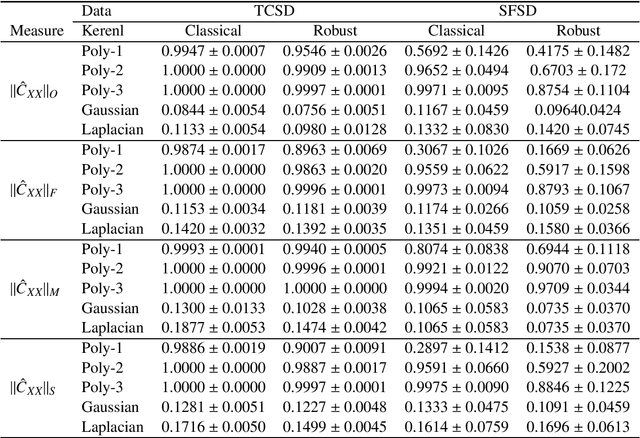
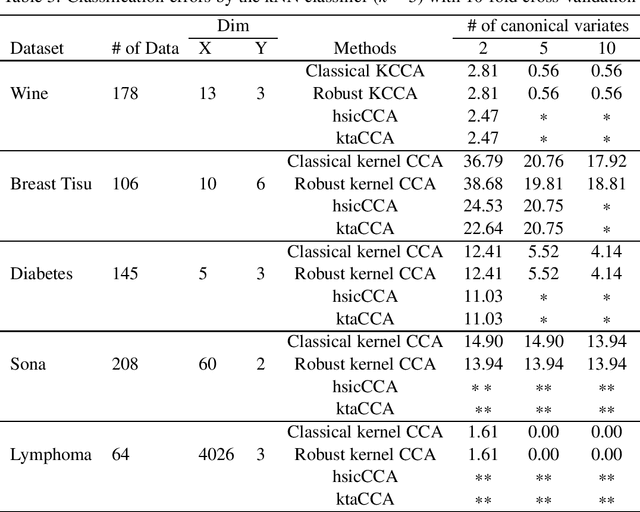
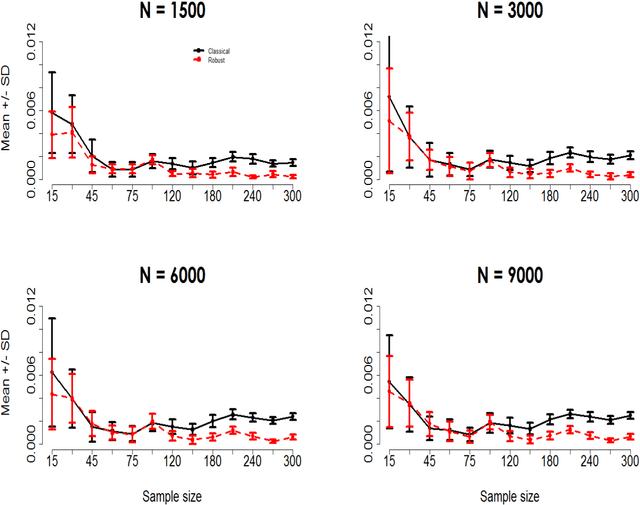
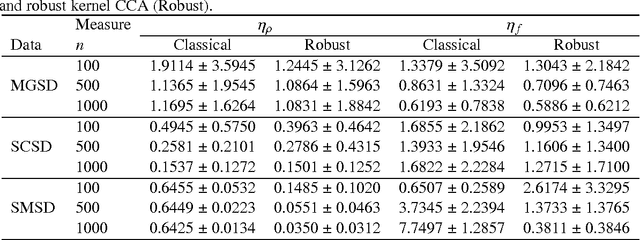
To the best of our knowledge, there are no general well-founded robust methods for statistical unsupervised learning. Most of the unsupervised methods explicitly or implicitly depend on the kernel covariance operator (kernel CO) or kernel cross-covariance operator (kernel CCO). They are sensitive to contaminated data, even when using bounded positive definite kernels. First, we propose robust kernel covariance operator (robust kernel CO) and robust kernel crosscovariance operator (robust kernel CCO) based on a generalized loss function instead of the quadratic loss function. Second, we propose influence function of classical kernel canonical correlation analysis (classical kernel CCA). Third, using this influence function, we propose a visualization method to detect influential observations from two sets of data. Finally, we propose a method based on robust kernel CO and robust kernel CCO, called robust kernel CCA, which is designed for contaminated data and less sensitive to noise than classical kernel CCA. The principles we describe also apply to many kernel methods which must deal with the issue of kernel CO or kernel CCO. Experiments on synthesized and imaging genetics analysis demonstrate that the proposed visualization and robust kernel CCA can be applied effectively to both ideal data and contaminated data. The robust methods show the superior performance over the state-of-the-art methods.