 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSynthesizable by Design: A Retrosynthesis-Guided Framework for Molecular Analog Generation
Jul 03, 2025The disconnect between AI-generated molecules with desirable properties and their synthetic feasibility remains a critical bottleneck in computational drug and material discovery. While generative AI has accelerated the proposal of candidate molecules, many of these structures prove challenging or impossible to synthesize using established chemical reactions. Here, we introduce SynTwins, a novel retrosynthesis-guided molecular analog design framework that designs synthetically accessible molecular analogs by emulating expert chemist strategies through a three-step process: retrosynthesis, similar building block searching, and virtual synthesis. In comparative evaluations, SynTwins demonstrates superior performance in generating synthetically accessible analogs compared to state-of-the-art machine learning models while maintaining high structural similarity to original target molecules. Furthermore, when integrated with existing molecule optimization frameworks, our hybrid approach produces synthetically feasible molecules with property profiles comparable to unconstrained molecule generators, yet its synthesizability ensured. Our comprehensive benchmarking across diverse molecular datasets demonstrates that SynTwins effectively bridges the gap between computational design and experimental synthesis, providing a practical solution for accelerating the discovery of synthesizable molecules with desired properties for a wide range of applications.
Predicting Chemical Reaction Outcomes Based on Electron Movements Using Machine Learning
Mar 13, 2025


Accurately predicting chemical reaction outcomes and potential byproducts is a fundamental task of modern chemistry, enabling the efficient design of synthetic pathways and driving progress in chemical science. Reaction mechanism, which tracks electron movements during chemical reactions, is critical for understanding reaction kinetics and identifying unexpected products. Here, we present Reactron, the first electron-based machine learning model for general reaction prediction. Reactron integrates electron movement into its predictions, generating detailed arrow-pushing diagrams that elucidate each mechanistic step leading to product formation. We demonstrate the high predictive performance of Reactron over existing product-only models by a large-scale reaction outcome prediction benchmark, and the adaptability of the model to learn new reactivity upon providing a few examples. Furthermore, it explores combinatorial reaction spaces, uncovering novel reactivities beyond its training data. With robust performance in both in- and out-of-distribution predictions, Reactron embodies human-like reasoning in chemistry and opens new frontiers in reaction discovery and synthesis design.
Assessing the Extrapolation Capability of Template-Free Retrosynthesis Models
Feb 29, 2024



Despite the acknowledged capability of template-free models in exploring unseen reaction spaces compared to template-based models for retrosynthesis prediction, their ability to venture beyond established boundaries remains relatively uncharted. In this study, we empirically assess the extrapolation capability of state-of-the-art template-free models by meticulously assembling an extensive set of out-of-distribution (OOD) reactions. Our findings demonstrate that while template-free models exhibit potential in predicting precursors with novel synthesis rules, their top-10 exact-match accuracy in OOD reactions is strikingly modest (< 1%). Furthermore, despite the capability of generating novel reactions, our investigation highlights a recurring issue where more than half of the novel reactions predicted by template-free models are chemically implausible. Consequently, we advocate for the future development of template-free models that integrate considerations of chemical feasibility when navigating unexplored regions of reaction space.
Inverse design of crystals using generalized invertible crystallographic representation
May 15, 2020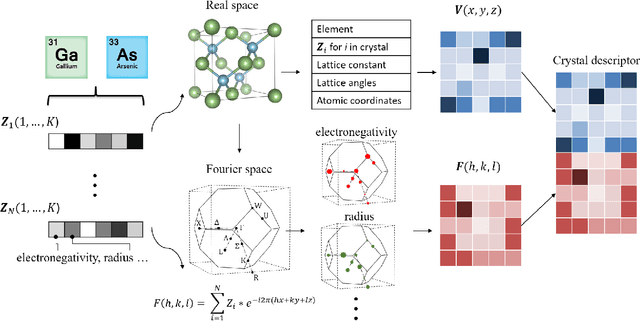
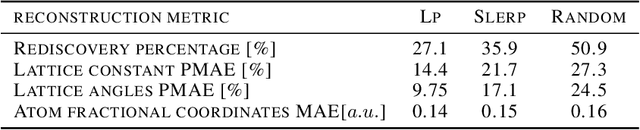
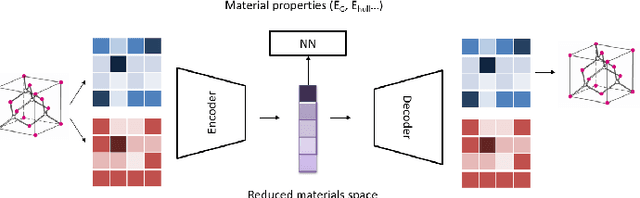
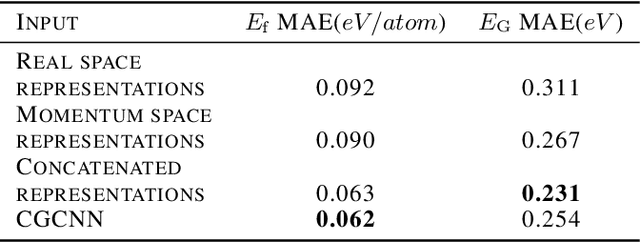
Deep learning has fostered many novel applications in materials informatics. However, the inverse design of inorganic crystals, $\textit{i.e.}$ generating new crystal structure with targeted properties, remains a grand challenge. An important ingredient for such generative models is an invertible representation that accesses the full periodic table. This is challenging due to limited data availability and the complexity of 3D periodic crystal structures. In this paper, we present a generalized invertible representation that encodes the crystallographic information into the descriptors in both real space and reciprocal space. Combining with a generative variational autoencoder (VAE), a wide range of crystallographic structures and chemistries with desired properties can be inverse-designed. We show that our VAE model predicts novel crystal structures that do not exist in the training and test database (Materials Project) with targeted formation energies and band gaps. We validate those predicted crystals by first-principles calculations. Finally, to design solids with practical applications, we address the sparse label problem by building a semi-supervised VAE and demonstrate its successful prediction of unique thermoelectric materials
Catalyst design using actively learned machine with non-ab initio input features towards CO2 reduction reactions
Sep 14, 2017
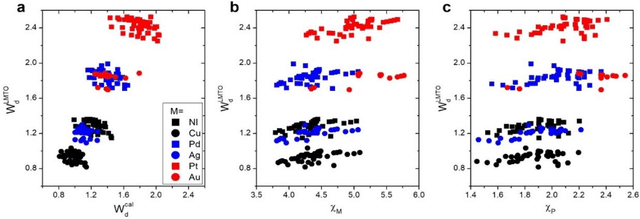
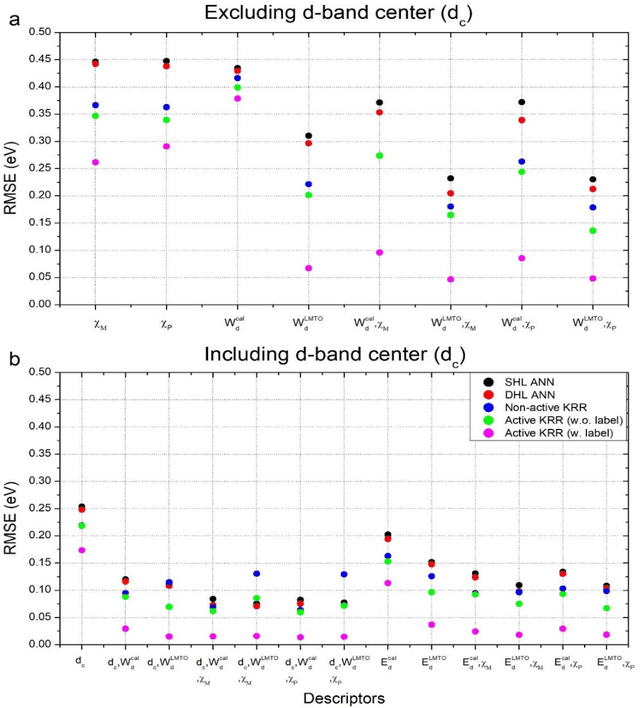
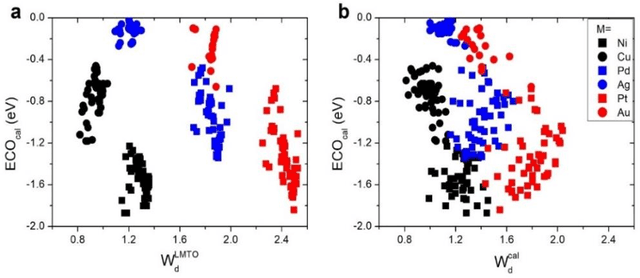
In conventional chemisorption model, the d-band center theory (augmented sometimes with the upper edge of d-band for imporved accuarcy) plays a central role in predicting adsorption energies and catalytic activity as a function of d-band center of the solid surfaces, but it requires density functional calculations that can be quite costly for large scale screening purposes of materials. In this work, we propose to use the d-band width of the muffin-tin orbital theory (to account for local coordination environment) plus electronegativity (to account for adsorbate renormalization) as a simple set of alternative descriptors for chemisorption, which do not demand the ab initio calculations. This pair of descriptors are then combined with machine learning methods, namely, artificial neural network (ANN) and kernel ridge regression (KRR), to allow large scale materials screenings. We show, for a toy set of 263 alloy systems, that the CO adsorption energy can be predicted with a remarkably small mean absolute deviation error of 0.05 eV, a significantly improved result as compared to 0.13 eV obtained with descriptors including costly d-band center calculations in literature. We achieved this high accuracy by utilizing an active learning algorithm, without which the accuracy was 0.18 eV otherwise. As a practical application of this machine, we identified Cu3Y@Cu as a highly active and cost-effective electrochemical CO2 reduction catalyst to produce CO with the overpotential 0.37 V lower than Au catalyst.