 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFast and Scalable Learning of Sparse Changes in High-Dimensional Gaussian Graphical Model Structure
May 23, 2018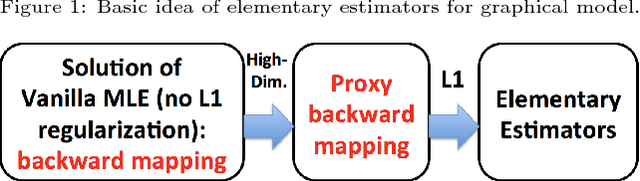
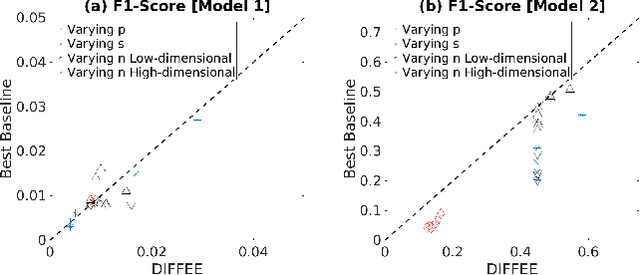
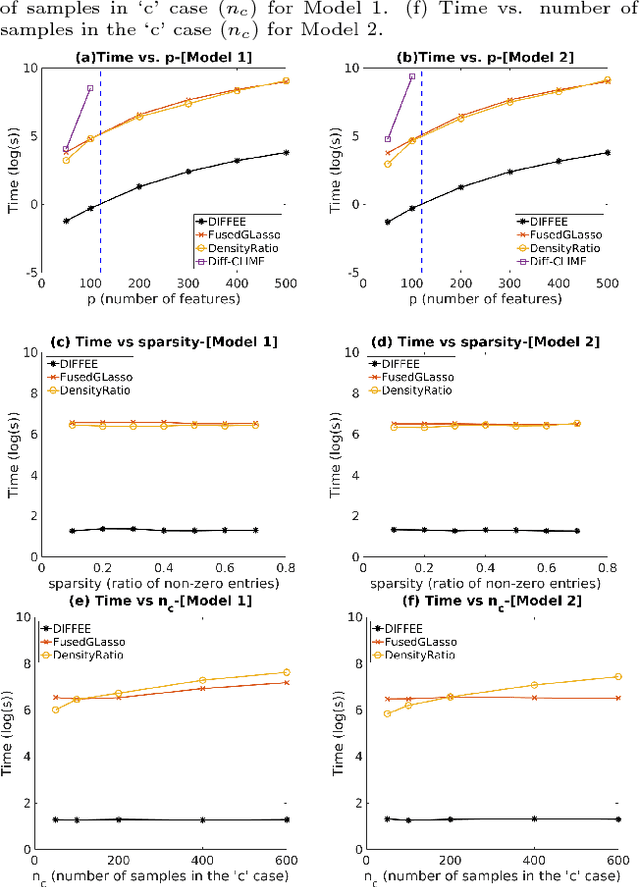
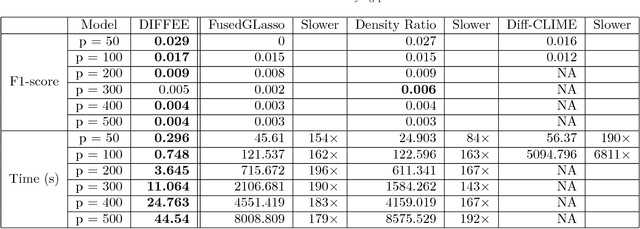
We focus on the problem of estimating the change in the dependency structures of two $p$-dimensional Gaussian Graphical models (GGMs). Previous studies for sparse change estimation in GGMs involve expensive and difficult non-smooth optimization. We propose a novel method, DIFFEE for estimating DIFFerential networks via an Elementary Estimator under a high-dimensional situation. DIFFEE is solved through a faster and closed form solution that enables it to work in large-scale settings. We conduct a rigorous statistical analysis showing that surprisingly DIFFEE achieves the same asymptotic convergence rates as the state-of-the-art estimators that are much more difficult to compute. Our experimental results on multiple synthetic datasets and one real-world data about brain connectivity show strong performance improvements over baselines, as well as significant computational benefits.
Black-box Generation of Adversarial Text Sequences to Evade Deep Learning Classifiers
May 23, 2018
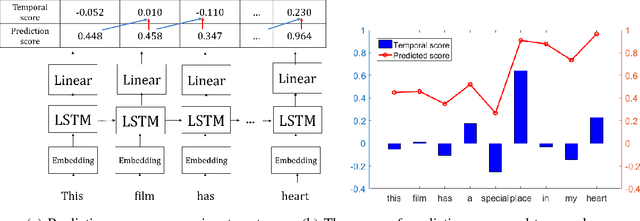
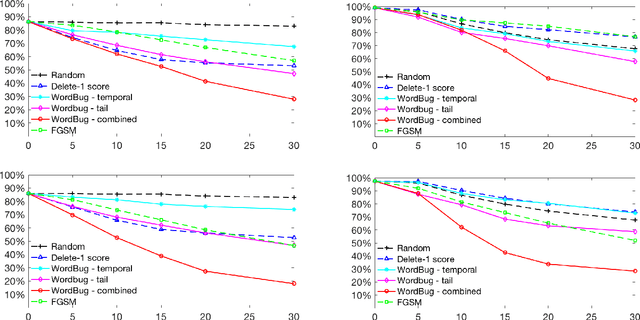
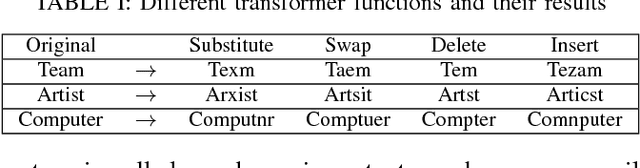
Although various techniques have been proposed to generate adversarial samples for white-box attacks on text, little attention has been paid to black-box attacks, which are more realistic scenarios. In this paper, we present a novel algorithm, DeepWordBug, to effectively generate small text perturbations in a black-box setting that forces a deep-learning classifier to misclassify a text input. We employ novel scoring strategies to identify the critical tokens that, if modified, cause the classifier to make an incorrect prediction. Simple character-level transformations are applied to the highest-ranked tokens in order to minimize the edit distance of the perturbation, yet change the original classification. We evaluated DeepWordBug on eight real-world text datasets, including text classification, sentiment analysis, and spam detection. We compare the result of DeepWordBug with two baselines: Random (Black-box) and Gradient (White-box). Our experimental results indicate that DeepWordBug reduces the prediction accuracy of current state-of-the-art deep-learning models, including a decrease of 68\% on average for a Word-LSTM model and 48\% on average for a Char-CNN model.
A Fast and Scalable Joint Estimator for Learning Multiple Related Sparse Gaussian Graphical Models
Mar 20, 2018
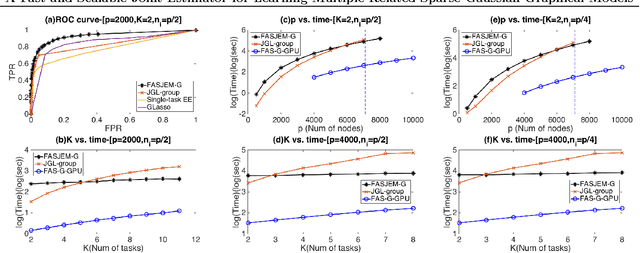
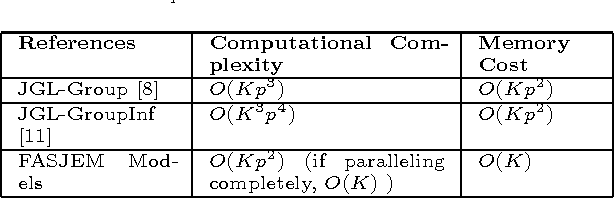
Estimating multiple sparse Gaussian Graphical Models (sGGMs) jointly for many related tasks (large $K$) under a high-dimensional (large $p$) situation is an important task. Most previous studies for the joint estimation of multiple sGGMs rely on penalized log-likelihood estimators that involve expensive and difficult non-smooth optimizations. We propose a novel approach, FASJEM for \underline{fa}st and \underline{s}calable \underline{j}oint structure-\underline{e}stimation of \underline{m}ultiple sGGMs at a large scale. As the first study of joint sGGM using the Elementary Estimator framework, our work has three major contributions: (1) We solve FASJEM through an entry-wise manner which is parallelizable. (2) We choose a proximal algorithm to optimize FASJEM. This improves the computational efficiency from $O(Kp^3)$ to $O(Kp^2)$ and reduces the memory requirement from $O(Kp^2)$ to $O(K)$. (3) We theoretically prove that FASJEM achieves a consistent estimation with a convergence rate of $O(\log(Kp)/n_{tot})$. On several synthetic and four real-world datasets, FASJEM shows significant improvements over baselines on accuracy, computational complexity, and memory costs.
Feature Squeezing: Detecting Adversarial Examples in Deep Neural Networks
Dec 05, 2017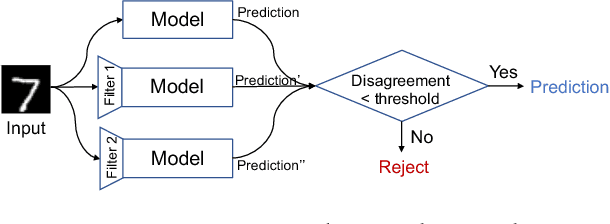

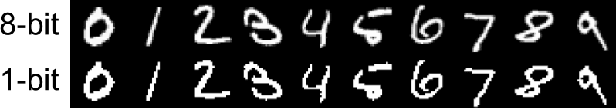

Although deep neural networks (DNNs) have achieved great success in many tasks, they can often be fooled by \emph{adversarial examples} that are generated by adding small but purposeful distortions to natural examples. Previous studies to defend against adversarial examples mostly focused on refining the DNN models, but have either shown limited success or required expensive computation. We propose a new strategy, \emph{feature squeezing}, that can be used to harden DNN models by detecting adversarial examples. Feature squeezing reduces the search space available to an adversary by coalescing samples that correspond to many different feature vectors in the original space into a single sample. By comparing a DNN model's prediction on the original input with that on squeezed inputs, feature squeezing detects adversarial examples with high accuracy and few false positives. This paper explores two feature squeezing methods: reducing the color bit depth of each pixel and spatial smoothing. These simple strategies are inexpensive and complementary to other defenses, and can be combined in a joint detection framework to achieve high detection rates against state-of-the-art attacks.
Prototype Matching Networks for Large-Scale Multi-label Genomic Sequence Classification
Nov 10, 2017
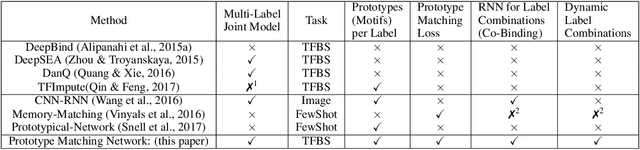
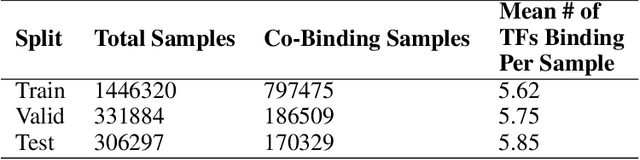
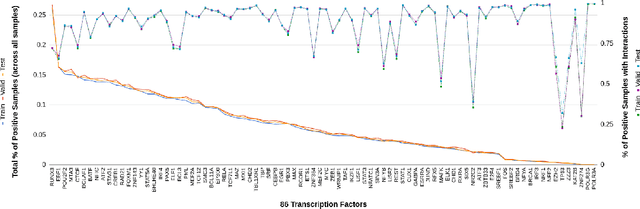
One of the fundamental tasks in understanding genomics is the problem of predicting Transcription Factor Binding Sites (TFBSs). With more than hundreds of Transcription Factors (TFs) as labels, genomic-sequence based TFBS prediction is a challenging multi-label classification task. There are two major biological mechanisms for TF binding: (1) sequence-specific binding patterns on genomes known as "motifs" and (2) interactions among TFs known as co-binding effects. In this paper, we propose a novel deep architecture, the Prototype Matching Network (PMN) to mimic the TF binding mechanisms. Our PMN model automatically extracts prototypes ("motif"-like features) for each TF through a novel prototype-matching loss. Borrowing ideas from few-shot matching models, we use the notion of support set of prototypes and an LSTM to learn how TFs interact and bind to genomic sequences. On a reference TFBS dataset with $2.1$ $million$ genomic sequences, PMN significantly outperforms baselines and validates our design choices empirically. To our knowledge, this is the first deep learning architecture that introduces prototype learning and considers TF-TF interactions for large-scale TFBS prediction. Not only is the proposed architecture accurate, but it also models the underlying biology.
Attend and Predict: Understanding Gene Regulation by Selective Attention on Chromatin
Nov 07, 2017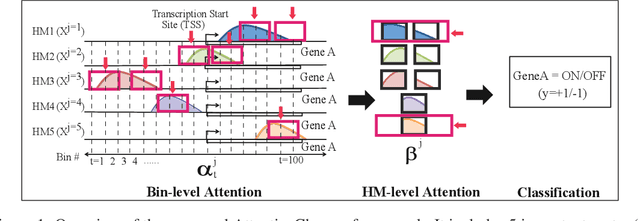
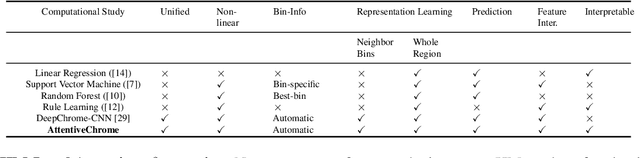
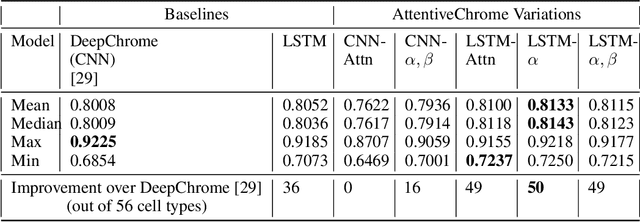
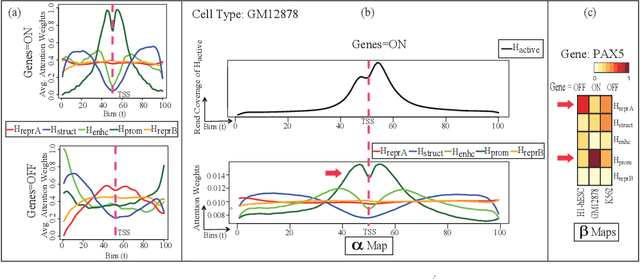
The past decade has seen a revolution in genomic technologies that enable a flood of genome-wide profiling of chromatin marks. Recent literature tried to understand gene regulation by predicting gene expression from large-scale chromatin measurements. Two fundamental challenges exist for such learning tasks: (1) genome-wide chromatin signals are spatially structured, high-dimensional and highly modular; and (2) the core aim is to understand what are the relevant factors and how they work together? Previous studies either failed to model complex dependencies among input signals or relied on separate feature analysis to explain the decisions. This paper presents an attention-based deep learning approach; we call AttentiveChrome, that uses a unified architecture to model and to interpret dependencies among chromatin factors for controlling gene regulation. AttentiveChrome uses a hierarchy of multiple Long short-term memory (LSTM) modules to encode the input signals and to model how various chromatin marks cooperate automatically. AttentiveChrome trains two levels of attention jointly with the target prediction, enabling it to attend differentially to relevant marks and to locate important positions per mark. We evaluate the model across 56 different cell types (tasks) in human. Not only is the proposed architecture more accurate, but its attention scores also provide a better interpretation than state-of-the-art feature visualization methods such as saliency map. Code and data are shared at www.deepchrome.org
A Theoretical Framework for Robustness of (Deep) Classifiers against Adversarial Examples
Sep 27, 2017
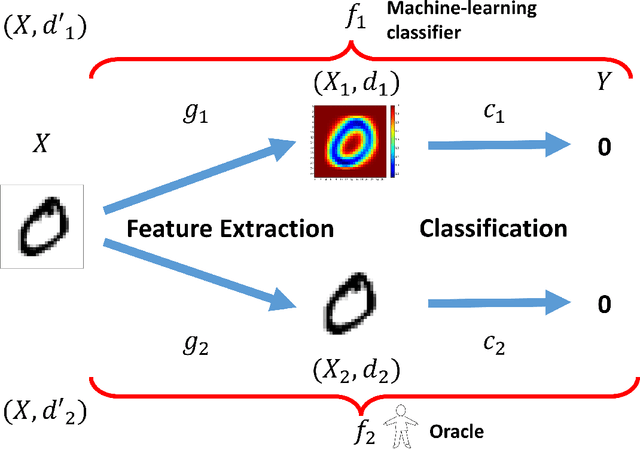
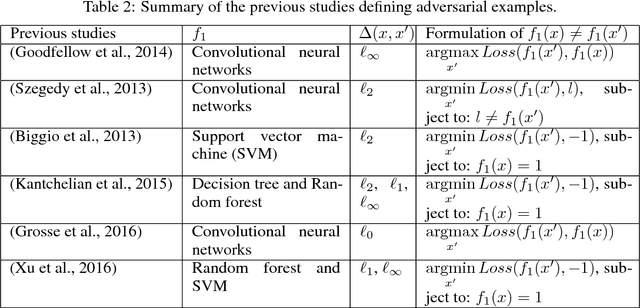
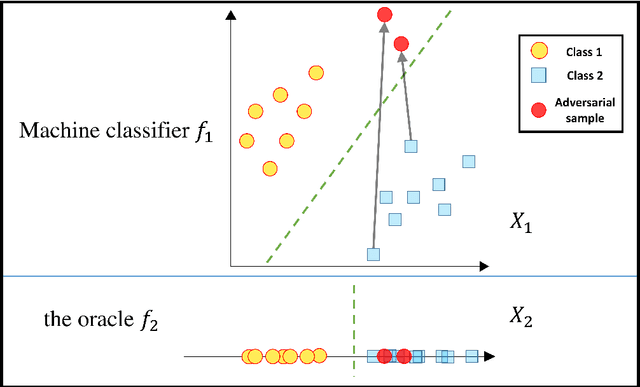
Most machine learning classifiers, including deep neural networks, are vulnerable to adversarial examples. Such inputs are typically generated by adding small but purposeful modifications that lead to incorrect outputs while imperceptible to human eyes. The goal of this paper is not to introduce a single method, but to make theoretical steps towards fully understanding adversarial examples. By using concepts from topology, our theoretical analysis brings forth the key reasons why an adversarial example can fool a classifier ($f_1$) and adds its oracle ($f_2$, like human eyes) in such analysis. By investigating the topological relationship between two (pseudo)metric spaces corresponding to predictor $f_1$ and oracle $f_2$, we develop necessary and sufficient conditions that can determine if $f_1$ is always robust (strong-robust) against adversarial examples according to $f_2$. Interestingly our theorems indicate that just one unnecessary feature can make $f_1$ not strong-robust, and the right feature representation learning is the key to getting a classifier that is both accurate and strong-robust.
A Constrained, Weighted-L1 Minimization Approach for Joint Discovery of Heterogeneous Neural Connectivity Graphs
Sep 21, 2017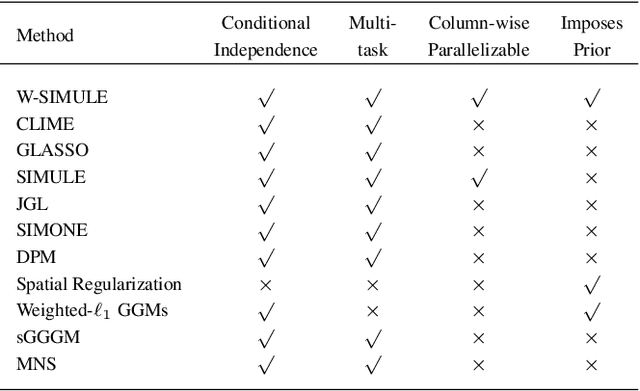
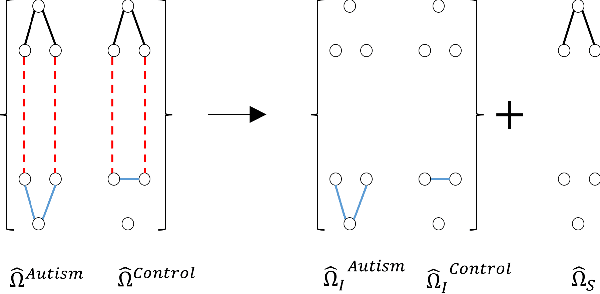
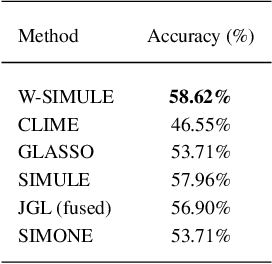
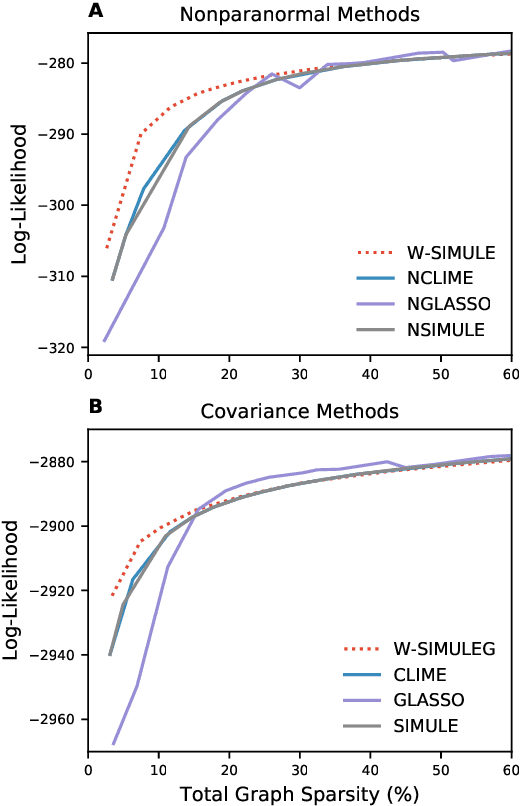
Determining functional brain connectivity is crucial to understanding the brain and neural differences underlying disorders such as autism. Recent studies have used Gaussian graphical models to learn brain connectivity via statistical dependencies across brain regions from neuroimaging. However, previous studies often fail to properly incorporate priors tailored to neuroscience, such as preferring shorter connections. To remedy this problem, the paper here introduces a novel, weighted-$\ell_1$, multi-task graphical model (W-SIMULE). This model elegantly incorporates a flexible prior, along with a parallelizable formulation. Additionally, W-SIMULE extends the often-used Gaussian assumption, leading to considerable performance increases. Here, applications to fMRI data show that W-SIMULE succeeds in determining functional connectivity in terms of (1) log-likelihood, (2) finding edges that differentiate groups, and (3) classifying different groups based on their connectivity, achieving 58.6\% accuracy on the ABIDE dataset. Having established W-SIMULE's effectiveness, it links four key areas to autism, all of which are consistent with the literature. Due to its elegant domain adaptivity, W-SIMULE can be readily applied to various data types to effectively estimate connectivity.
GaKCo: a Fast GApped k-mer string Kernel using COunting
Sep 18, 2017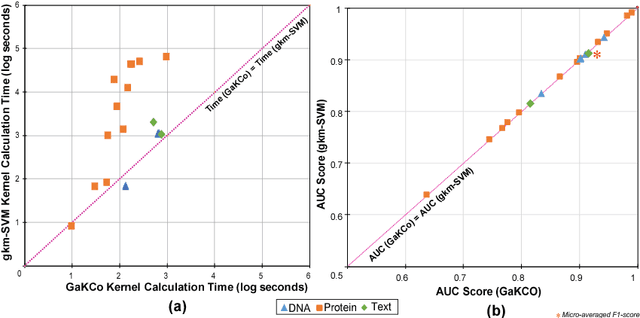
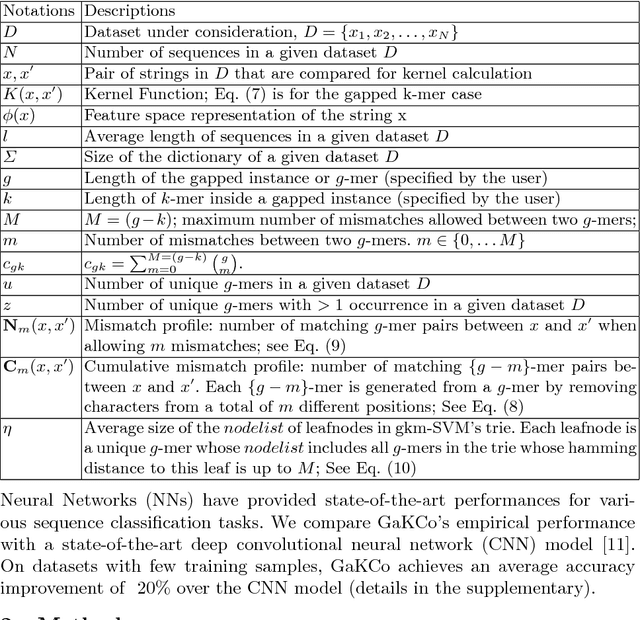
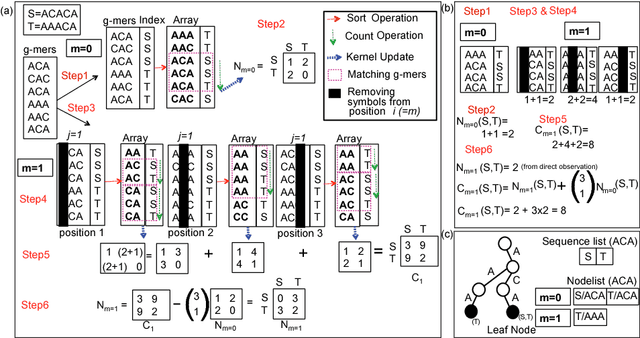
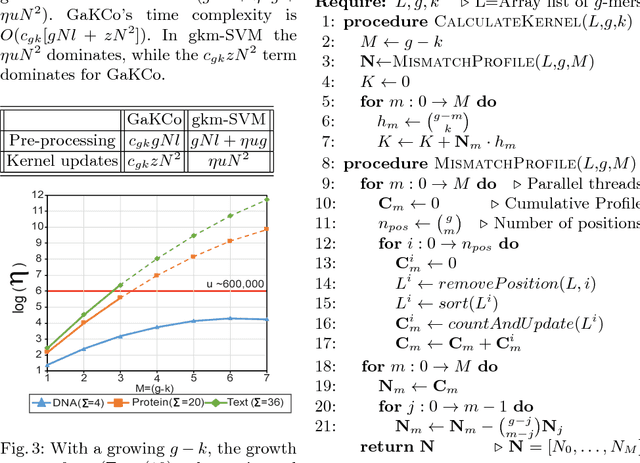
String Kernel (SK) techniques, especially those using gapped $k$-mers as features (gk), have obtained great success in classifying sequences like DNA, protein, and text. However, the state-of-the-art gk-SK runs extremely slow when we increase the dictionary size ($\Sigma$) or allow more mismatches ($M$). This is because current gk-SK uses a trie-based algorithm to calculate co-occurrence of mismatched substrings resulting in a time cost proportional to $O(\Sigma^{M})$. We propose a \textbf{fast} algorithm for calculating \underline{Ga}pped $k$-mer \underline{K}ernel using \underline{Co}unting (GaKCo). GaKCo uses associative arrays to calculate the co-occurrence of substrings using cumulative counting. This algorithm is fast, scalable to larger $\Sigma$ and $M$, and naturally parallelizable. We provide a rigorous asymptotic analysis that compares GaKCo with the state-of-the-art gk-SK. Theoretically, the time cost of GaKCo is independent of the $\Sigma^{M}$ term that slows down the trie-based approach. Experimentally, we observe that GaKCo achieves the same accuracy as the state-of-the-art and outperforms its speed by factors of 2, 100, and 4, on classifying sequences of DNA (5 datasets), protein (12 datasets), and character-based English text (2 datasets), respectively. GaKCo is shared as an open source tool at \url{https://github.com/QData/GaKCo-SVM}
A constrained L1 minimization approach for estimating multiple Sparse Gaussian or Nonparanormal Graphical Models
Sep 18, 2017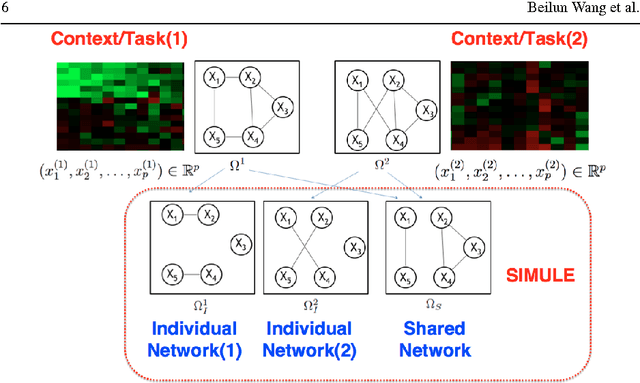

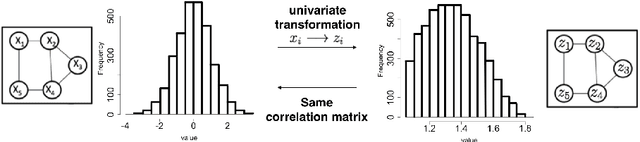
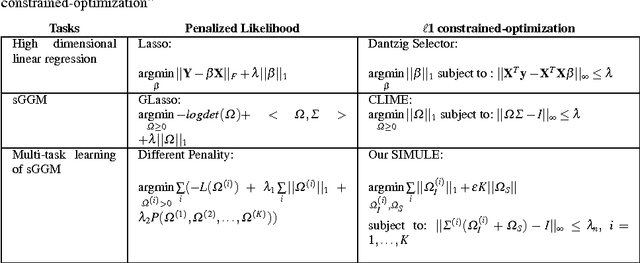
Identifying context-specific entity networks from aggregated data is an important task, arising often in bioinformatics and neuroimaging. Computationally, this task can be formulated as jointly estimating multiple different, but related, sparse Undirected Graphical Models (UGM) from aggregated samples across several contexts. Previous joint-UGM studies have mostly focused on sparse Gaussian Graphical Models (sGGMs) and can't identify context-specific edge patterns directly. We, therefore, propose a novel approach, SIMULE (detecting Shared and Individual parts of MULtiple graphs Explicitly) to learn multi-UGM via a constrained L1 minimization. SIMULE automatically infers both specific edge patterns that are unique to each context and shared interactions preserved among all the contexts. Through the L1 constrained formulation, this problem is cast as multiple independent subtasks of linear programming that can be solved efficiently in parallel. In addition to Gaussian data, SIMULE can also handle multivariate Nonparanormal data that greatly relaxes the normality assumption that many real-world applications do not follow. We provide a novel theoretical proof showing that SIMULE achieves a consistent result at the rate O(log(Kp)/n_{tot}). On multiple synthetic datasets and two biomedical datasets, SIMULE shows significant improvement over state-of-the-art multi-sGGM and single-UGM baselines.