 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRelate and Predict: Structure-Aware Prediction with Jointly Optimized Neural DAG
Mar 03, 2021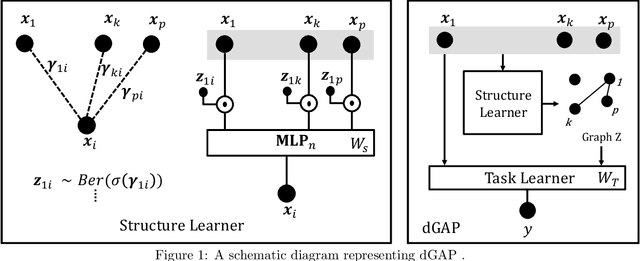
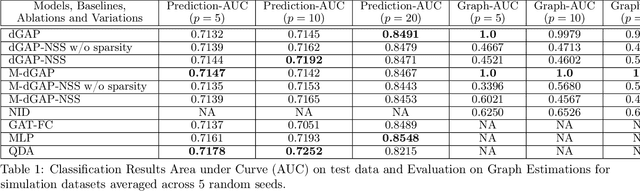
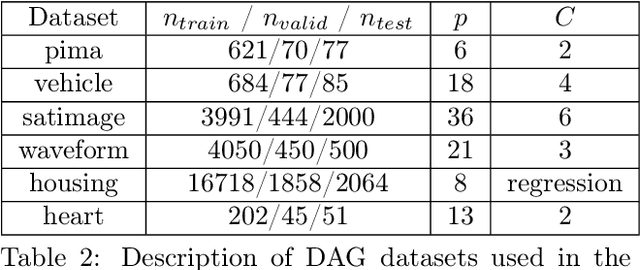
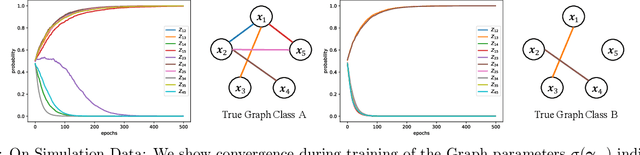
Understanding relationships between feature variables is one important way humans use to make decisions. However, state-of-the-art deep learning studies either focus on task-agnostic statistical dependency learning or do not model explicit feature dependencies during prediction. We propose a deep neural network framework, dGAP, to learn neural dependency Graph and optimize structure-Aware target Prediction simultaneously. dGAP trains towards a structure self-supervision loss and a target prediction loss jointly. Our method leads to an interpretable model that can disentangle sparse feature relationships, informing the user how relevant dependencies impact the target task. We empirically evaluate dGAP on multiple simulated and real datasets. dGAP is not only more accurate, but can also recover correct dependency structure.
General Multi-label Image Classification with Transformers
Nov 27, 2020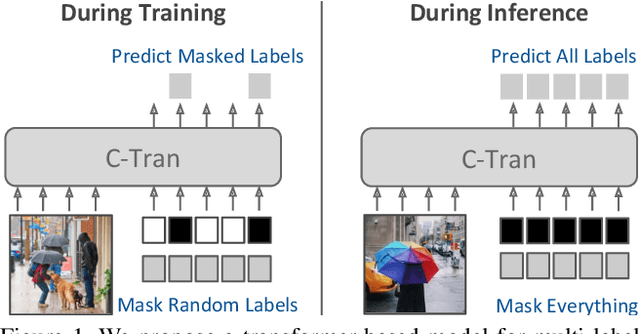
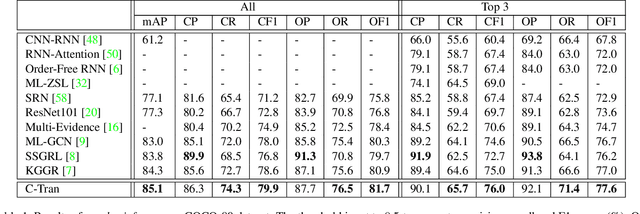


Multi-label image classification is the task of predicting a set of labels corresponding to objects, attributes or other entities present in an image. In this work we propose the Classification Transformer (C-Tran), a general framework for multi-label image classification that leverages Transformers to exploit the complex dependencies among visual features and labels. Our approach consists of a Transformer encoder trained to predict a set of target labels given an input set of masked labels, and visual features from a convolutional neural network. A key ingredient of our method is a label mask training objective that uses a ternary encoding scheme to represent the state of the labels as positive, negative, or unknown during training. Our model shows state-of-the-art performance on challenging datasets such as COCO and Visual Genome. Moreover, because our model explicitly represents the uncertainty of labels during training, it is more general by allowing us to produce improved results for images with partial or extra label annotations during inference. We demonstrate this additional capability in the COCO, Visual Genome, News500, and CUB image datasets.
Measuring Visual Generalization in Continuous Control from Pixels
Oct 13, 2020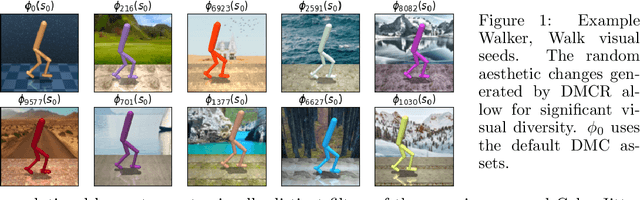
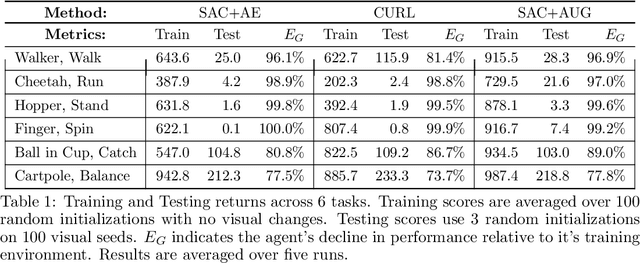
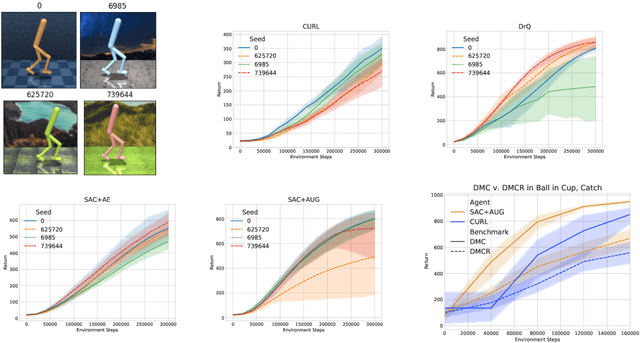
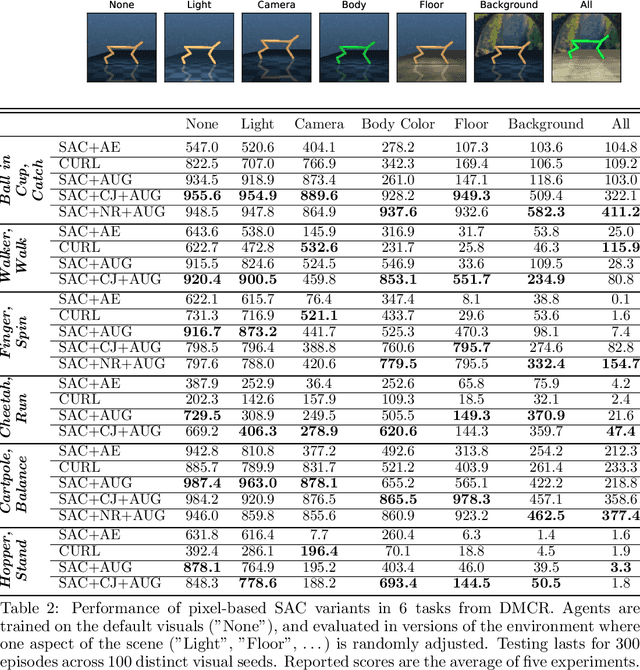
Self-supervised learning and data augmentation have significantly reduced the performance gap between state and image-based reinforcement learning agents in continuous control tasks. However, it is still unclear whether current techniques can face a variety of visual conditions required by real-world environments. We propose a challenging benchmark that tests agents' visual generalization by adding graphical variety to existing continuous control domains. Our empirical analysis shows that current methods struggle to generalize across a diverse set of visual changes, and we examine the specific factors of variation that make these tasks difficult. We find that data augmentation techniques outperform self-supervised learning approaches and that more significant image transformations provide better visual generalization \footnote{The benchmark and our augmented actor-critic implementation are open-sourced @ https://github.com/jakegrigsby/dmc_remastered)
Searching for a Search Method: Benchmarking Search Algorithms for Generating NLP Adversarial Examples
Oct 12, 2020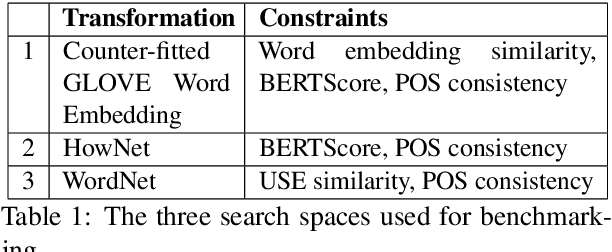
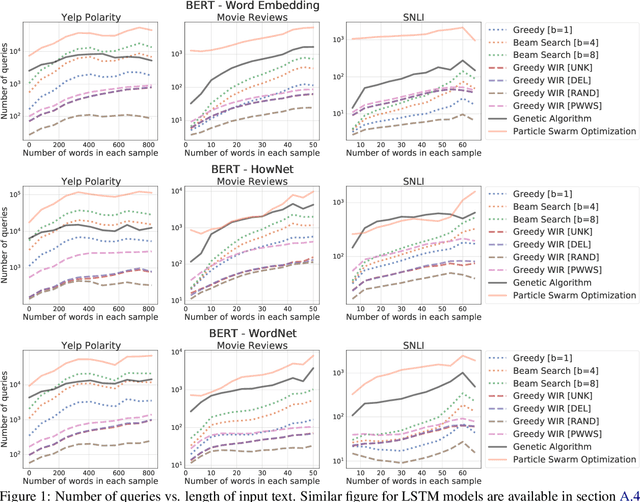
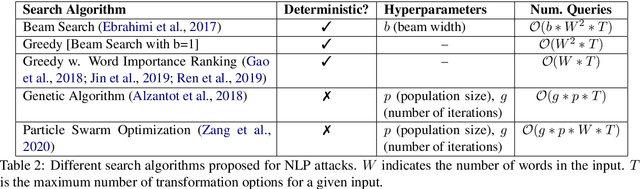
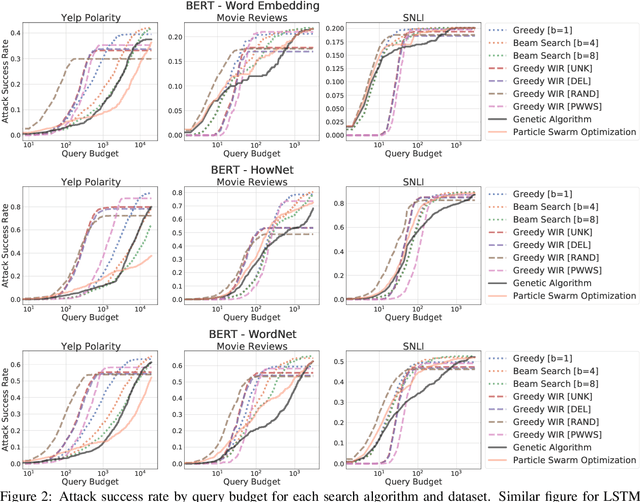
We study the behavior of several black-box search algorithms used for generating adversarial examples for natural language processing (NLP) tasks. We perform a fine-grained analysis of three elements relevant to search: search algorithm, search space, and search budget. When new search algorithms are proposed in past work, the attack search space is often modified alongside the search algorithm. Without ablation studies benchmarking the search algorithm change with the search space held constant, one cannot tell if an increase in attack success rate is a result of an improved search algorithm or a less restrictive search space. Additionally, many previous studies fail to properly consider the search algorithms' run-time cost, which is essential for downstream tasks like adversarial training. Our experiments provide a reproducible benchmark of search algorithms across a variety of search spaces and query budgets to guide future research in adversarial NLP. Based on our experiments, we recommend greedy attacks with word importance ranking when under a time constraint or attacking long inputs, and either beam search or particle swarm optimization otherwise. Code implementation shared via https://github.com/QData/TextAttack-Search-Benchmark
TextAttack: A Framework for Adversarial Attacks in Natural Language Processing
May 13, 2020
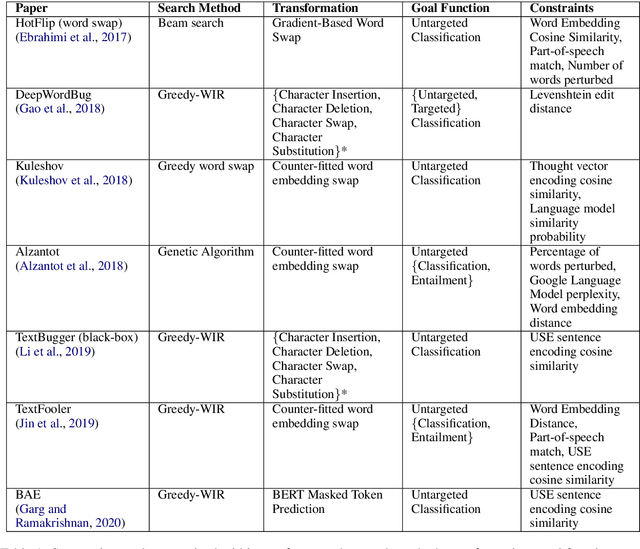
TextAttack is a library for running adversarial attacks against natural language processing (NLP) models. TextAttack builds attacks from four components: a search method, goal function, transformation, and a set of constraints. Researchers can use these components to easily assemble new attacks. Individual components can be isolated and compared for easier ablation studies. TextAttack currently supports attacks on models trained for text classification and entailment across a variety of datasets. Additionally, TextAttack's modular design makes it easily extensible to new NLP tasks, models, and attack strategies. TextAttack code and tutorials are available at https://github.com/QData/TextAttack.
Reevaluating Adversarial Examples in Natural Language
Apr 25, 2020
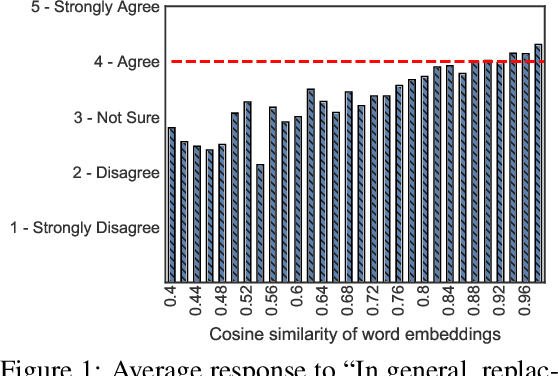
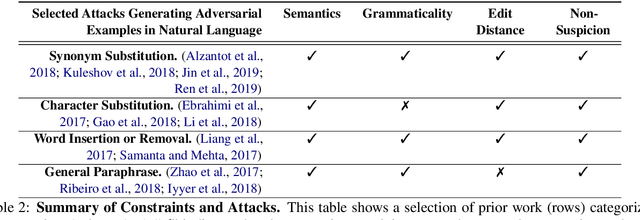
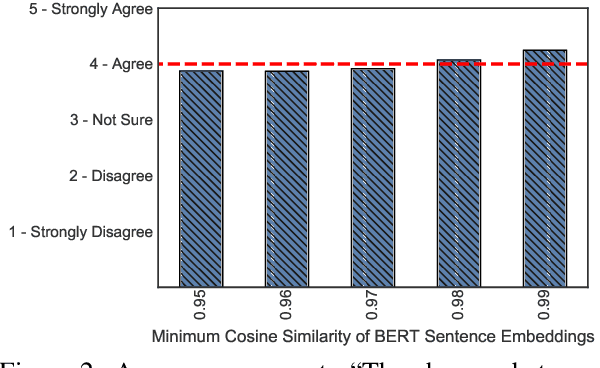
State-of-the-art attacks on NLP models have different definitions of what constitutes a successful attack. These differences make the attacks difficult to compare. We propose to standardize definitions of natural language adversarial examples based on a set of linguistic constraints: semantics, grammaticality, edit distance, and non-suspicion. We categorize previous attacks based on these constraints. For each constraint, we suggest options for human and automatic evaluation methods. We use these methods to evaluate two state-of-the-art synonym substitution attacks. We find that perturbations often do not preserve semantics, and 45\% introduce grammatical errors. Next, we conduct human studies to find a threshold for each evaluation method that aligns with human judgment. Human surveys reveal that to truly preserve semantics, we need to significantly increase the minimum cosine similarity between the embeddings of swapped words and sentence encodings of original and perturbed inputs. After tightening these constraints to agree with the judgment of our human annotators, the attacks produce valid, successful adversarial examples. But quality comes at a cost: attack success rate drops by over 70 percentage points. Finally, we introduce TextAttack, a library for adversarial attacks in NLP.
Differential Network Learning Beyond Data Samples
Apr 24, 2020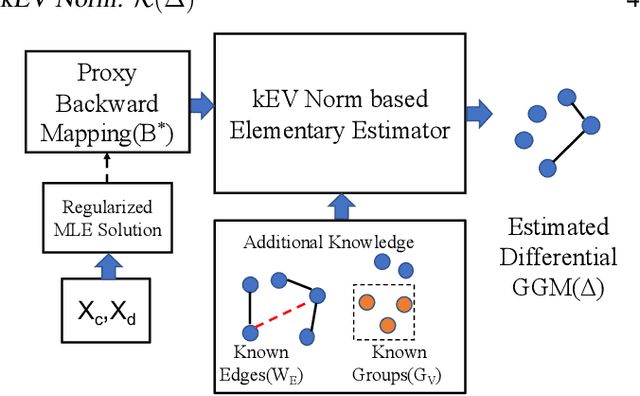
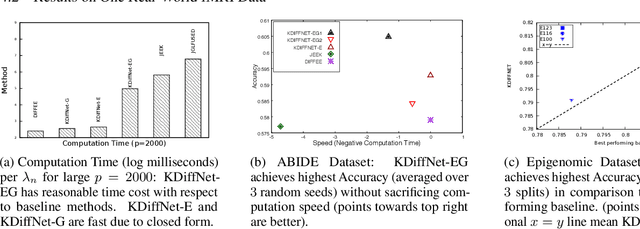
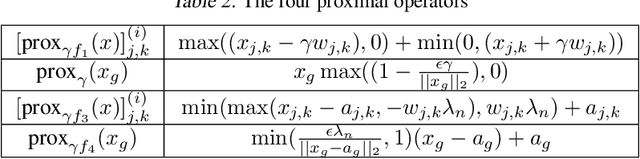
Learning the change of statistical dependencies between random variables is an essential task for many real-life applications, mostly in the high dimensional low sample regime. In this paper, we propose a novel differential parameter estimator that, in comparison to current methods, simultaneously allows (a) the flexible integration of multiple sources of information (data samples, variable groupings, extra pairwise evidence, etc.), (b) being scalable to a large number of variables, and (c) achieving a sharp asymptotic convergence rate. Our experiments, on more than 100 simulated and two real-world datasets, validate the flexibility of our approach and highlight the benefits of integrating spatial and anatomic information for brain connectome change discovery and epigenetic network identification.
Curriculum Labeling: Self-paced Pseudo-Labeling for Semi-Supervised Learning
Jan 16, 2020
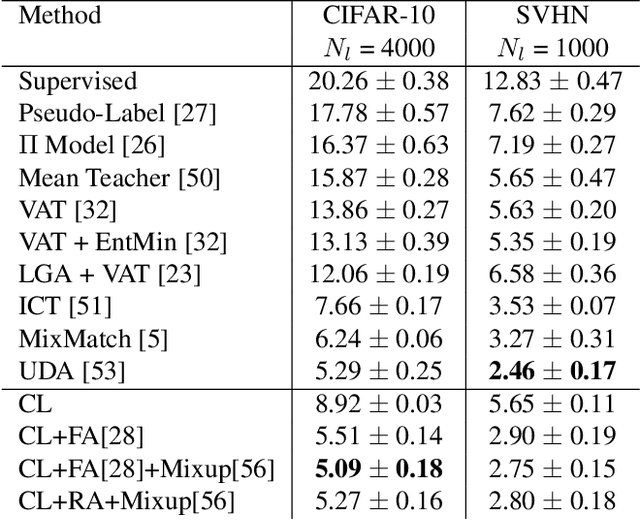
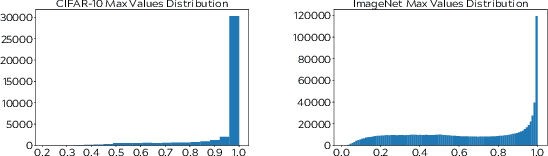
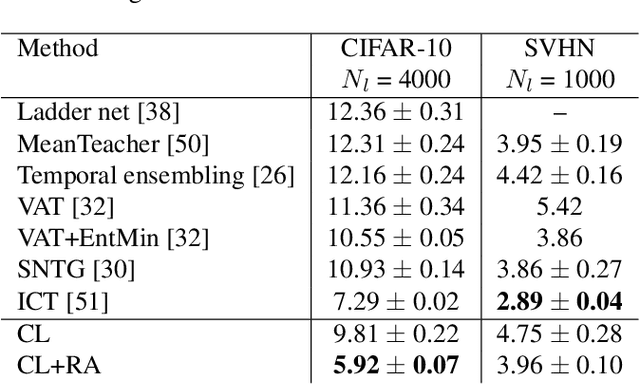
Semi-supervised learning aims to take advantage of a large amount of unlabeled data to improve the accuracy of a model that only has access to a small number of labeled examples. We propose curriculum labeling, an approach that exploits pseudo-labeling for propagating labels to unlabeled samples in an iterative and self-paced fashion. This approach is surprisingly simple and effective and surpasses or is comparable with the best methods proposed in the recent literature across all the standard benchmarks for image classification. Notably, we obtain 94.91% accuracy on CIFAR-10 using only 4,000 labeled samples, and 88.56% top-5 accuracy on Imagenet-ILSVRC using 128,000 labeled samples. In contrast to prior works, our approach shows improvements even in a more realistic scenario that leverages out-of-distribution unlabeled data samples.
Neural Message Passing for Multi-Label Classification
Apr 17, 2019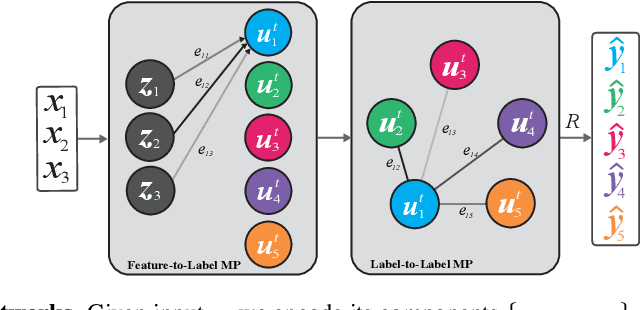
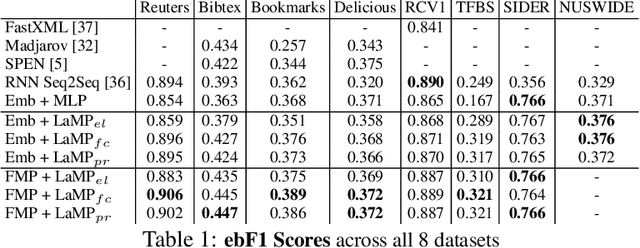
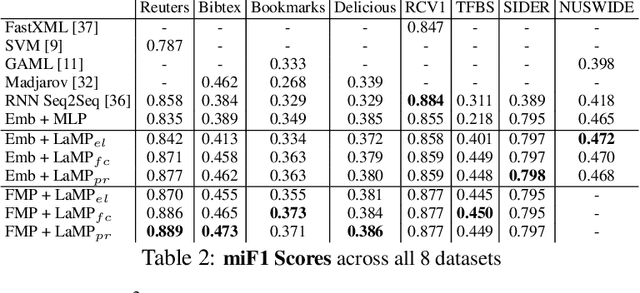
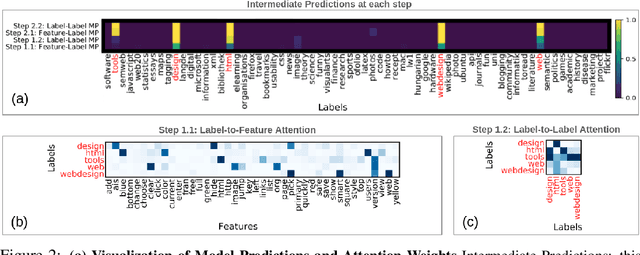
Multi-label classification (MLC) is the task of assigning a set of target labels for a given sample. Modeling the combinatorial label interactions in MLC has been a long-haul challenge. We propose Label Message Passing (LaMP) Neural Networks to efficiently model the joint prediction of multiple labels. LaMP treats labels as nodes on a label-interaction graph and computes the hidden representation of each label node conditioned on the input using attention-based neural message passing. Attention enables LaMP to assign different importance to neighbor nodes per label, learning how labels interact (implicitly). The proposed models are simple, accurate, interpretable, structure-agnostic, and applicable for predicting dense labels since LaMP is incredibly parallelizable. We validate the benefits of LaMP on seven real-world MLC datasets, covering a broad spectrum of input/output types and outperforming the state-of-the-art results. Notably, LaMP enables intuitive interpretation of how classifying each label depends on the elements of a sample and at the same time rely on its interaction with other labels. We provide our code and datasets at https://github.com/QData/LaMP
A Fast and Scalable Joint Estimator for Integrating Additional Knowledge in Learning Multiple Related Sparse Gaussian Graphical Models
Jul 17, 2018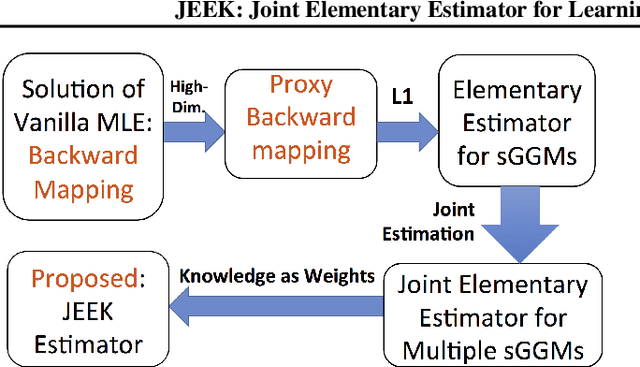
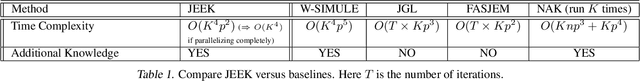
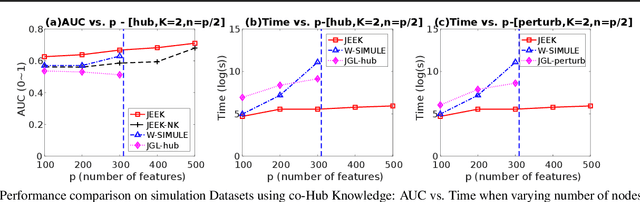
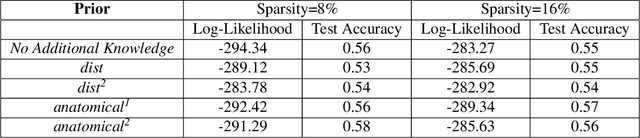
We consider the problem of including additional knowledge in estimating sparse Gaussian graphical models (sGGMs) from aggregated samples, arising often in bioinformatics and neuroimaging applications. Previous joint sGGM estimators either fail to use existing knowledge or cannot scale-up to many tasks (large $K$) under a high-dimensional (large $p$) situation. In this paper, we propose a novel \underline{J}oint \underline{E}lementary \underline{E}stimator incorporating additional \underline{K}nowledge (JEEK) to infer multiple related sparse Gaussian Graphical models from large-scale heterogeneous data. Using domain knowledge as weights, we design a novel hybrid norm as the minimization objective to enforce the superposition of two weighted sparsity constraints, one on the shared interactions and the other on the task-specific structural patterns. This enables JEEK to elegantly consider various forms of existing knowledge based on the domain at hand and avoid the need to design knowledge-specific optimization. JEEK is solved through a fast and entry-wise parallelizable solution that largely improves the computational efficiency of the state-of-the-art $O(p^5K^4)$ to $O(p^2K^4)$. We conduct a rigorous statistical analysis showing that JEEK achieves the same convergence rate $O(\log(Kp)/n_{tot})$ as the state-of-the-art estimators that are much harder to compute. Empirically, on multiple synthetic datasets and two real-world data, JEEK outperforms the speed of the state-of-arts significantly while achieving the same level of prediction accuracy. Available as R tool "jeek"