 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentic multi-fidelity learning of quasiparticle and excitonic properties
Jun 05, 2026Many-body GW-Bethe-Salpeter equation calculations are essential for accurate simulations of electronic structure and optical properties in modern low-dimensional nanomaterials. However, these methods are computationally demanding and can exhibit localized numerical instabilities or convergence failures that are difficult to detect within high-throughput workflows. We introduce an agent-guided multi-fidelity framework for correcting GW-Bethe-Salpeter excited-state landscapes in strained MoS2-WS2 bilayers. Across stacking registries, strain branches and reciprocal-space samplings, the workflow identifies spike-like excursions, near-zero-gap collapse and cross-fidelity inconsistencies associated with fragile long-wavelength dielectric screening. A structural agent evaluates calculations by assigning confidence weights and selectively using a small number of high-accuracy reference points. Machine learning models then transfer information across related systems and apply Gaussian process corrections to recover improved quasiparticle gaps and exciton binding energies, with calibrated uncertainty estimates. The approach corrects numerically induced artifacts without erasing physical strain dependence and substantially improves agreement with higher-fidelity references relative to a no-agent baseline. These results show that reliable surrogate learning for excited-state materials requires explicit diagnosis of numerical fragility, not direct interpolation of raw first-principles data points. The proposed framework is readily transferable to other optoelectronic nanomaterials characterized by strong quantum confinement, such as quantum dots, nanoribbons, layered two-dimensional semiconductors, and hybrid perovskite nanostructures.
Deep Generative Learning of Magnetic Frustration in Artificial Spin Ice from Magnetic Force Microscopy Images
Jul 23, 2025Increasingly large datasets of microscopic images with atomic resolution facilitate the development of machine learning methods to identify and analyze subtle physical phenomena embedded within the images. In this work, microscopic images of honeycomb lattice spin-ice samples serve as datasets from which we automate the calculation of net magnetic moments and directional orientations of spin-ice configurations. In the first stage of our workflow, machine learning models are trained to accurately predict magnetic moments and directions within spin-ice structures. Variational Autoencoders (VAEs), an emergent unsupervised deep learning technique, are employed to generate high-quality synthetic magnetic force microscopy (MFM) images and extract latent feature representations, thereby reducing experimental and segmentation errors. The second stage of proposed methodology enables precise identification and prediction of frustrated vertices and nanomagnetic segments, effectively correlating structural and functional aspects of microscopic images. This facilitates the design of optimized spin-ice configurations with controlled frustration patterns, enabling potential on-demand synthesis.
Teacher-student training improves accuracy and efficiency of machine learning inter-atomic potentials
Feb 07, 2025



Machine learning inter-atomic potentials (MLIPs) are revolutionizing the field of molecular dynamics (MD) simulations. Recent MLIPs have tended towards more complex architectures trained on larger datasets. The resulting increase in computational and memory costs may prohibit the application of these MLIPs to perform large-scale MD simulations. Here, we present a teacher-student training framework in which the latent knowledge from the teacher (atomic energies) is used to augment the students' training. We show that the light-weight student MLIPs have faster MD speeds at a fraction of the memory footprint compared to the teacher models. Remarkably, the student models can even surpass the accuracy of the teachers, even though both are trained on the same quantum chemistry dataset. Our work highlights a practical method for MLIPs to reduce the resources required for large-scale MD simulations.
Automated discovery of a robust interatomic potential for aluminum
Mar 10, 2020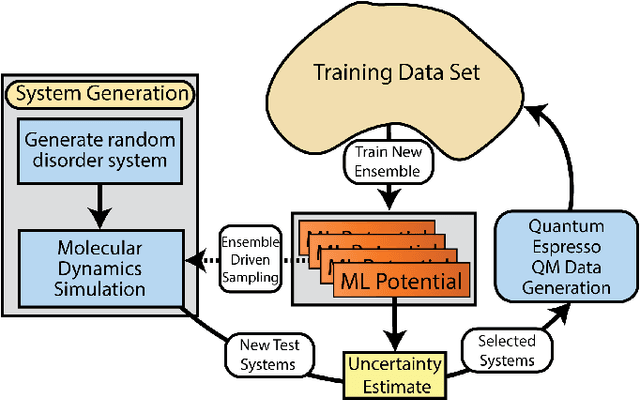
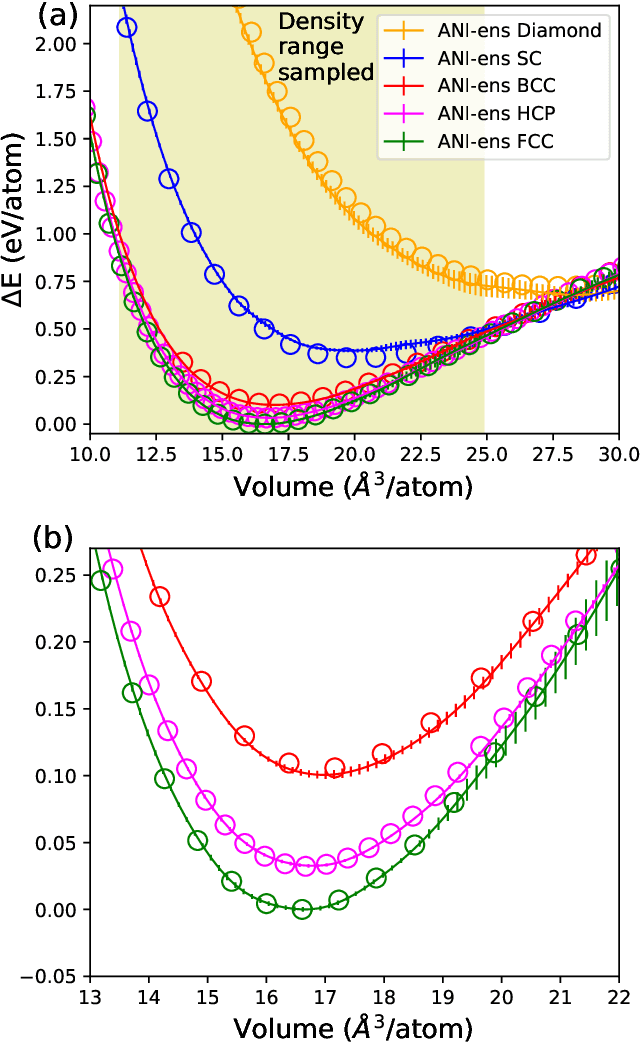
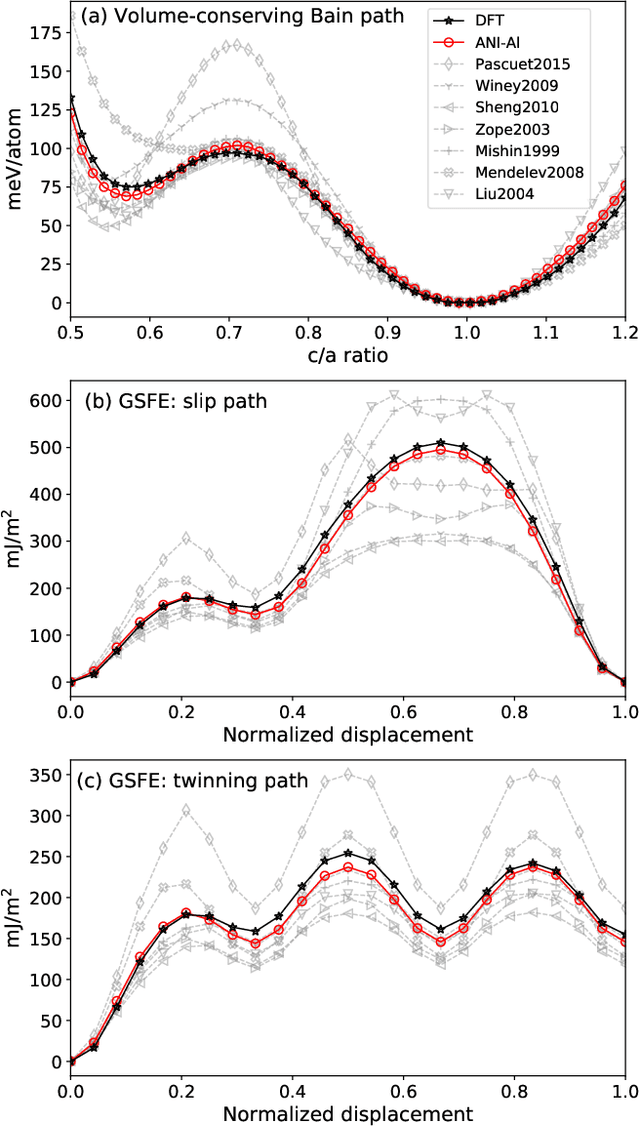
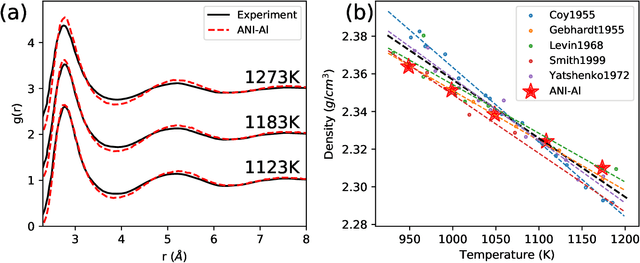
Atomistic molecular dynamics simulation is an important tool for predicting materials properties. Accuracy depends crucially on the model for the interatomic potential. The gold standard would be quantum mechanics (QM) based force calculations, but such a first-principles approach becomes prohibitively expensive at large system sizes. Efficient machine learning models (ML) have become increasingly popular as surrogates for QM. Neural networks with many thousands of parameters excel in capturing structure within a large dataset, but may struggle to extrapolate beyond the scope of the available data. Here we present a highly automated active learning approach to iteratively collect new QM data that best resolves weaknesses in the existing ML model. We exemplify our approach by developing a general potential for elemental aluminum. At each active learning iteration, the method (1) trains an ANI-style neural network potential from the available data, (2) uses this potential to drive molecular dynamics simulations, and (3) collects new QM data whenever the neural network identifies an atomic configuration for which it cannot make a good prediction. All molecular dynamics simulations are initialized to a disordered configuration, and then driven according to randomized, time-varying temperatures. This nonequilibrium molecular dynamics forms a variety of crystalline and defected configurations. By training on all such automatically collected data, we produce ANI-Al, our new interatomic potential for aluminum. We demonstrate the remarkable transferability of ANI-Al by benchmarking against experimental data, e.g., the radial distribution function in melt, various properties of the stable face-centered cubic (FCC) crystal, and the coexistence curve between melt and FCC.