 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCritical Windows of Complexity Control: When Transformers Decide to Reason or Memorize
May 06, 2026Recent work has shown that Transformers' compositional generalization is governed by \emph{complexity control}, initialization scale and weight decay, which steers training toward low-complexity reasoning solutions rather than high-complexity memorization. Existing analyses, however, treat complexity control as a single static hyperparameter choice, leaving open \emph{when} during training this control is actually decisive. We show that the memorization-versus-reasoning fate of a Transformer is determined within a sharp, identifiable window of training. On a controlled compositional task we find that (i)~weight decay applied for a single 25\%-of-training window matches full-training weight decay in out-of-distribution (OOD) accuracy ($0.93$ vs $0.91$); (ii)~holding total regularization budget constant, placing it in the middle of training yields $5{-}9\times$ higher OOD accuracy than placing it early; (iii)~the boundary of the critical window is remarkably sharp, window onset shifted by as little as $100$ optimization steps causes mean OOD to jump from chance ($0.15$) to reasoning-regime ($0.61$); (iv)~the window's position depends systematically on initialization scale, but the basin of attraction for reasoning solutions \emph{shrinks} at small initialization, contradicting the prevailing recommendation that smaller initialization is uniformly better. We further show that the critical-window phenomenon is task-specific: it does not appear on grokking with modular arithmetic, where properly tuned constant weight decay matches scheduled weight decay.
Multi-Scale Reversible Chaos Game Representation: A Unified Framework for Sequence Classification
Apr 20, 2026Biological classification with interpretability remains a challenging task. For this, we introduce a novel encoding framework, Multi-Scale Reversible Chaos Game Representation (MS-RCGR), that transforms biological sequences into multi-resolution geometric representations with guaranteed reversibility. Unlike traditional sequence encoding methods, MS-RCGR employs rational arithmetic and hierarchical k-mer decomposition to generate scale-invariant features that preserve complete sequence information while enabling diverse analytical approaches. Our framework bridges three distinct paradigms for sequence analysis: (1) traditional machine learning using extracted geometric features, (2) computer vision models operating on CGR-generated images, and (3) hybrid approaches combining protein language model embeddings with CGR features. Through comprehensive experiments on synthetic DNA and protein datasets encompassing seven distinct sequence classes, we demonstrate that MS-RCGR features consistently enhance classification performance across all paradigms. Notably, our hybrid approach combining pre-trained language model embeddings (ESM2, ProtT5) with MS-RCGR features achieves superior performance compared to either method alone. The reversibility property of our encoding ensures no information loss during transformation, while multi-scale analysis captures patterns ranging from individual nucleotides to complex motif structures. Our results indicate that MS-RCGR provides a flexible, interpretable, and high-performing foundation for biological sequence analysis.
DPSR: Differentially Private Sparse Reconstruction via Multi-Stage Denoising for Recommender Systems
Dec 22, 2025



Differential privacy (DP) has emerged as the gold standard for protecting user data in recommender systems, but existing privacy-preserving mechanisms face a fundamental challenge: the privacy-utility tradeoff inevitably degrades recommendation quality as privacy budgets tighten. We introduce DPSR (Differentially Private Sparse Reconstruction), a novel three-stage denoising framework that fundamentally addresses this limitation by exploiting the inherent structure of rating matrices -- sparsity, low-rank properties, and collaborative patterns. DPSR consists of three synergistic stages: (1) \textit{information-theoretic noise calibration} that adaptively reduces noise for high-information ratings, (2) \textit{collaborative filtering-based denoising} that leverages item-item similarities to remove privacy noise, and (3) \textit{low-rank matrix completion} that exploits latent structure for signal recovery. Critically, all denoising operations occur \textit{after} noise injection, preserving differential privacy through the post-processing immunity theorem while removing both privacy-induced and inherent data noise. Through extensive experiments on synthetic datasets with controlled ground truth, we demonstrate that DPSR achieves 5.57\% to 9.23\% RMSE improvement over state-of-the-art Laplace and Gaussian mechanisms across privacy budgets ranging from $\varepsilon=0.1$ to $\varepsilon=10.0$ (all improvements statistically significant with $p < 0.05$, most $p < 0.001$). Remarkably, at $\varepsilon=1.0$, DPSR achieves RMSE of 0.9823, \textit{outperforming even the non-private baseline} (1.0983), demonstrating that our denoising pipeline acts as an effective regularizer that removes data noise in addition to privacy noise.
Boosting t-SNE Efficiency for Sequencing Data: Insights from Kernel Selection
Dec 17, 2025



Dimensionality reduction techniques are essential for visualizing and analyzing high-dimensional biological sequencing data. t-distributed Stochastic Neighbor Embedding (t-SNE) is widely used for this purpose, traditionally employing the Gaussian kernel to compute pairwise similarities. However, the Gaussian kernel's lack of data-dependence and computational overhead limit its scalability and effectiveness for categorical biological sequences. Recent work proposed the isolation kernel as an alternative, yet it may not optimally capture sequence similarities. In this study, we comprehensively evaluate nine different kernel functions for t-SNE applied to molecular sequences, using three embedding methods: One-Hot Encoding, Spike2Vec, and minimizers. Through both subjective visualization and objective metrics (including neighborhood preservation scores), we demonstrate that the cosine similarity kernel in general outperforms other kernels, including Gaussian and isolation kernels, achieving superior runtime efficiency and better preservation of pairwise distances in low-dimensional space. We further validate our findings through extensive classification and clustering experiments across six diverse biological datasets (Spike7k, Host, ShortRead, Rabies, Genome, and Breast Cancer), employing multiple machine learning algorithms and evaluation metrics. Our results show that kernel selection significantly impacts not only visualization quality but also downstream analytical tasks, with the cosine similarity kernel providing the most robust performance across different data types and embedding strategies, making it particularly suitable for large-scale biological sequence analysis.
Sequence-to-Image Transformation for Sequence Classification Using Rips Complex Construction and Chaos Game Representation
Dec 10, 2025Traditional feature engineering approaches for molecular sequence classification suffer from sparsity issues and computational complexity, while deep learning models often underperform on tabular biological data. This paper introduces a novel topological approach that transforms molecular sequences into images by combining Chaos Game Representation (CGR) with Rips complex construction from algebraic topology. Our method maps sequence elements to 2D coordinates via CGR, computes pairwise distances, and constructs Rips complexes to capture both local structural and global topological features. We provide formal guarantees on representation uniqueness, topological stability, and information preservation. Extensive experiments on anticancer peptide datasets demonstrate superior performance over vector-based, sequence language models, and existing image-based methods, achieving 86.8\% and 94.5\% accuracy on breast and lung cancer datasets, respectively. The topological representation preserves critical sequence information while enabling effective utilization of vision-based deep learning architectures for molecular sequence analysis.
Murmur2Vec: A Hashing Based Solution For Embedding Generation Of COVID-19 Spike Sequences
Dec 10, 2025Early detection and characterization of coronavirus disease (COVID-19), caused by SARS-CoV-2, remain critical for effective clinical response and public-health planning. The global availability of large-scale viral sequence data presents significant opportunities for computational analysis; however, existing approaches face notable limitations. Phylogenetic tree-based methods are computationally intensive and do not scale efficiently to today's multi-million-sequence datasets. Similarly, current embedding-based techniques often rely on aligned sequences or exhibit suboptimal predictive performance and high runtime costs, creating barriers to practical large-scale analysis. In this study, we focus on the most prevalent SARS-CoV-2 lineages associated with the spike protein region and introduce a scalable embedding method that leverages hashing to generate compact, low-dimensional representations of spike sequences. These embeddings are subsequently used to train a variety of machine learning models for supervised lineage classification. We conduct an extensive evaluation comparing our approach with multiple baseline and state-of-the-art biological sequence embedding methods across diverse metrics. Our results demonstrate that the proposed embeddings offer substantial improvements in efficiency, achieving up to 86.4\% classification accuracy while reducing embedding generation time by as much as 99.81\%. This highlights the method's potential as a fast, effective, and scalable solution for large-scale viral sequence analysis.
Robust Gradient Descent via Heavy-Ball Momentum with Predictive Extrapolation
Dec 10, 2025Accelerated gradient methods like Nesterov's Accelerated Gradient (NAG) achieve faster convergence on well-conditioned problems but often diverge on ill-conditioned or non-convex landscapes due to aggressive momentum accumulation. We propose Heavy-Ball Synthetic Gradient Extrapolation (HB-SGE), a robust first-order method that combines heavy-ball momentum with predictive gradient extrapolation. Unlike classical momentum methods that accumulate historical gradients, HB-SGE estimates future gradient directions using local Taylor approximations, providing adaptive acceleration while maintaining stability. We prove convergence guarantees for strongly convex functions and demonstrate empirically that HB-SGE prevents divergence on problems where NAG and standard momentum fail. On ill-conditioned quadratics (condition number $κ=50$), HB-SGE converges in 119 iterations while both SGD and NAG diverge. On the non-convex Rosenbrock function, HB-SGE achieves convergence in 2,718 iterations where classical momentum methods diverge within 10 steps. While NAG remains faster on well-conditioned problems, HB-SGE provides a robust alternative with speedup over SGD across diverse landscapes, requiring only $O(d)$ memory overhead and the same hyperparameters as standard momentum.
Sequence Analysis Using the Bezier Curve
Mar 18, 2025



The analysis of sequences (e.g., protein, DNA, and SMILES string) is essential for disease diagnosis, biomaterial engineering, genetic engineering, and drug discovery domains. Conventional analytical methods focus on transforming sequences into numerical representations for applying machine learning/deep learning-based sequence characterization. However, their efficacy is constrained by the intrinsic nature of deep learning (DL) models, which tend to exhibit suboptimal performance when applied to tabular data. An alternative group of methodologies endeavors to convert biological sequences into image forms by applying the concept of Chaos Game Representation (CGR). However, a noteworthy drawback of these methods lies in their tendency to map individual elements of the sequence onto a relatively small subset of designated pixels within the generated image. The resulting sparse image representation may not adequately encapsulate the comprehensive sequence information, potentially resulting in suboptimal predictions. In this study, we introduce a novel approach to transform sequences into images using the B\'ezier curve concept for element mapping. Mapping the elements onto a curve enhances the sequence information representation in the respective images, hence yielding better DL-based classification performance. We employed different sequence datasets to validate our system by using different classification tasks, and the results illustrate that our B\'ezier curve method is able to achieve good performance for all the tasks.
Converting Time Series Data to Numeric Representations Using Alphabetic Mapping and k-mer strategy
Dec 29, 2024

In the realm of data analysis and bioinformatics, representing time series data in a manner akin to biological sequences offers a novel approach to leverage sequence analysis techniques. Transforming time series signals into molecular sequence-type representations allows us to enhance pattern recognition by applying sophisticated sequence analysis techniques (e.g. $k$-mers based representation) developed in bioinformatics, uncovering hidden patterns and relationships in complex, non-linear time series data. This paper proposes a method to transform time series signals into biological/molecular sequence-type representations using a unique alphabetic mapping technique. By generating 26 ranges corresponding to the 26 letters of the English alphabet, each value within the time series is mapped to a specific character based on its range. This conversion facilitates the application of sequence analysis algorithms, typically used in bioinformatics, to analyze time series data. We demonstrate the effectiveness of this approach by converting real-world time series signals into character sequences and performing sequence classification. The resulting sequences can be utilized for various sequence-based analysis techniques, offering a new perspective on time series data representation and analysis.
Hilbert Curve Based Molecular Sequence Analysis
Dec 29, 2024
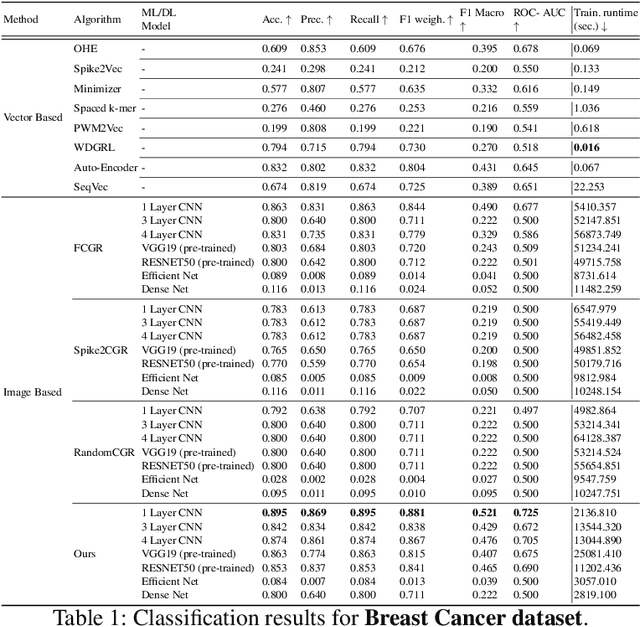

Accurate molecular sequence analysis is a key task in the field of bioinformatics. To apply molecular sequence classification algorithms, we first need to generate the appropriate representations of the sequences. Traditional numeric sequence representation techniques are mostly based on sequence alignment that faces limitations in the form of lack of accuracy. Although several alignment-free techniques have also been introduced, their tabular data form results in low performance when used with Deep Learning (DL) models compared to the competitive performance observed in the case of image-based data. To find a solution to this problem and to make Deep Learning (DL) models function to their maximum potential while capturing the important spatial information in the sequence data, we propose a universal Hibert curve-based Chaos Game Representation (CGR) method. This method is a transformative function that involves a novel Alphabetic index mapping technique used in constructing Hilbert curve-based image representation from molecular sequences. Our method can be globally applied to any type of molecular sequence data. The Hilbert curve-based image representations can be used as input to sophisticated vision DL models for sequence classification. The proposed method shows promising results as it outperforms current state-of-the-art methods by achieving a high accuracy of $94.5$\% and an F1 score of $93.9\%$ when tested with the CNN model on the lung cancer dataset. This approach opens up a new horizon for exploring molecular sequence analysis using image classification methods.