 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSequence-to-Image Transformation for Sequence Classification Using Rips Complex Construction and Chaos Game Representation
Dec 10, 2025Traditional feature engineering approaches for molecular sequence classification suffer from sparsity issues and computational complexity, while deep learning models often underperform on tabular biological data. This paper introduces a novel topological approach that transforms molecular sequences into images by combining Chaos Game Representation (CGR) with Rips complex construction from algebraic topology. Our method maps sequence elements to 2D coordinates via CGR, computes pairwise distances, and constructs Rips complexes to capture both local structural and global topological features. We provide formal guarantees on representation uniqueness, topological stability, and information preservation. Extensive experiments on anticancer peptide datasets demonstrate superior performance over vector-based, sequence language models, and existing image-based methods, achieving 86.8\% and 94.5\% accuracy on breast and lung cancer datasets, respectively. The topological representation preserves critical sequence information while enabling effective utilization of vision-based deep learning architectures for molecular sequence analysis.
Post-Training Non-Uniform Quantization for Convolutional Neural Networks
Dec 10, 2024



Despite the success of CNN models on a variety of Image classification and segmentation tasks, their extensive computational and storage demands pose considerable challenges for real-world deployment on resource constrained devices. Quantization is one technique that aims to alleviate these large storage requirements and speed up the inference process by reducing the precision of model parameters to lower-bit representations. In this paper, we introduce a novel post-training quantization method for model weights. Our method finds optimal clipping thresholds and scaling factors along with mathematical guarantees that our method minimizes quantization noise. Empirical results on Real World Datasets demonstrate that our quantization scheme significantly reduces model size and computational requirements while preserving model accuracy.
Impact Of Missing Data Imputation On The Fairness And Accuracy Of Graph Node Classifiers
Nov 01, 2022Analysis of the fairness of machine learning (ML) algorithms recently attracted many researchers' interest. Most ML methods show bias toward protected groups, which limits the applicability of ML models in many applications like crime rate prediction etc. Since the data may have missing values which, if not appropriately handled, are known to further harmfully affect fairness. Many imputation methods are proposed to deal with missing data. However, the effect of missing data imputation on fairness is not studied well. In this paper, we analyze the effect on fairness in the context of graph data (node attributes) imputation using different embedding and neural network methods. Extensive experiments on six datasets demonstrate severe fairness issues in missing data imputation under graph node classification. We also find that the choice of the imputation method affects both fairness and accuracy. Our results provide valuable insights into graph data fairness and how to handle missingness in graphs efficiently. This work also provides directions regarding theoretical studies on fairness in graph data.
Effective and scalable clustering of SARS-CoV-2 sequences
Sep 10, 2021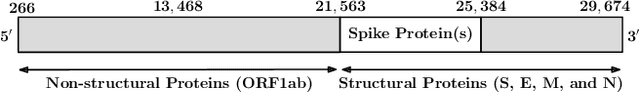
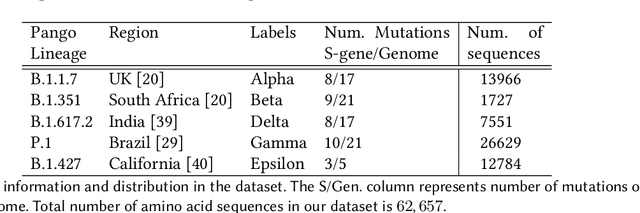
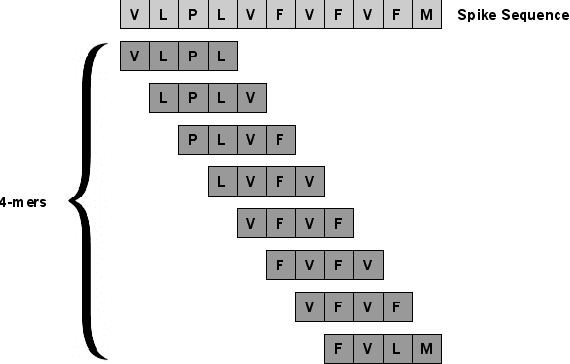
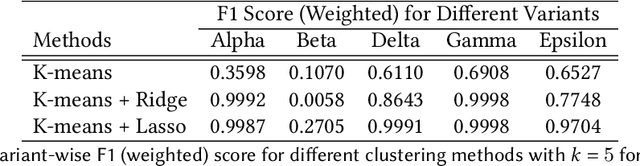
SARS-CoV-2, like any other virus, continues to mutate as it spreads, according to an evolutionary process. Unlike any other virus, the number of currently available sequences of SARS-CoV-2 in public databases such as GISAID is already several million. This amount of data has the potential to uncover the evolutionary dynamics of a virus like never before. However, a million is already several orders of magnitude beyond what can be processed by the traditional methods designed to reconstruct a virus's evolutionary history, such as those that build a phylogenetic tree. Hence, new and scalable methods will need to be devised in order to make use of the ever increasing number of viral sequences being collected. Since identifying variants is an important part of understanding the evolution of a virus, in this paper, we propose an approach based on clustering sequences to identify the current major SARS-CoV-2 variants. Using a $k$-mer based feature vector generation and efficient feature selection methods, our approach is effective in identifying variants, as well as being efficient and scalable to millions of sequences. Such a clustering method allows us to show the relative proportion of each variant over time, giving the rate of spread of each variant in different locations -- something which is important for vaccine development and distribution. We also compute the importance of each amino acid position of the spike protein in identifying a given variant in terms of information gain. Positions of high variant-specific importance tend to agree with those reported by the USA's Centers for Disease Control and Prevention (CDC), further demonstrating our approach.
Computing Graph Descriptors on Edge Streams
Sep 02, 2021
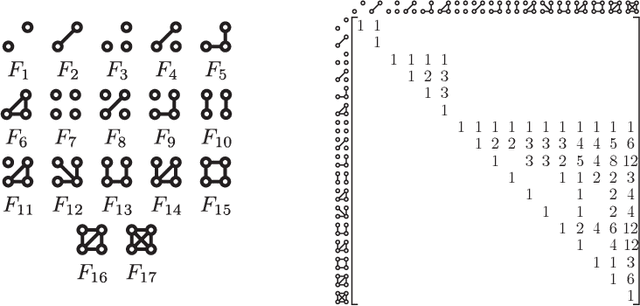
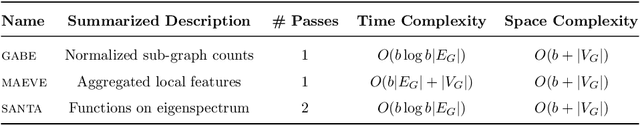
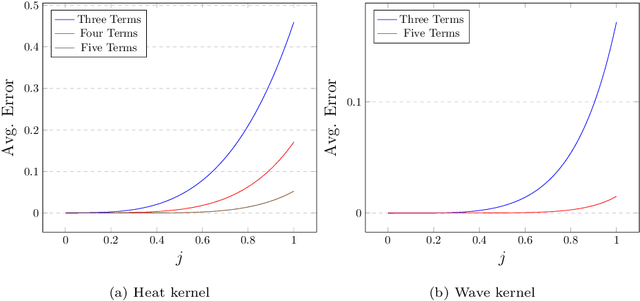
Graph feature extraction is a fundamental task in graphs analytics. Using feature vectors (graph descriptors) in tandem with data mining algorithms that operate on Euclidean data, one can solve problems such as classification, clustering, and anomaly detection on graph-structured data. This idea has proved fruitful in the past, with spectral-based graph descriptors providing state-of-the-art classification accuracy on benchmark datasets. However, these algorithms do not scale to large graphs since: 1) they require storing the entire graph in memory, and 2) the end-user has no control over the algorithm's runtime. In this paper, we present single-pass streaming algorithms to approximate structural features of graphs (counts of subgraphs of order $k \geq 4$). Operating on edge streams allows us to avoid keeping the entire graph in memory, and controlling the sample size enables us to control the time taken by the algorithm. We demonstrate the efficacy of our descriptors by analyzing the approximation error, classification accuracy, and scalability to massive graphs. Our experiments showcase the effect of the sample size on approximation error and predictive accuracy. The proposed descriptors are applicable on graphs with millions of edges within minutes and outperform the state-of-the-art descriptors in classification accuracy.
A k-mer Based Approach for SARS-CoV-2 Variant Identification
Aug 25, 2021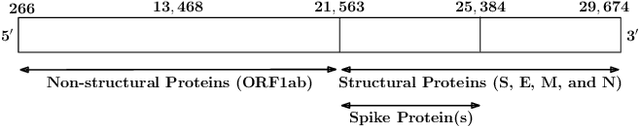
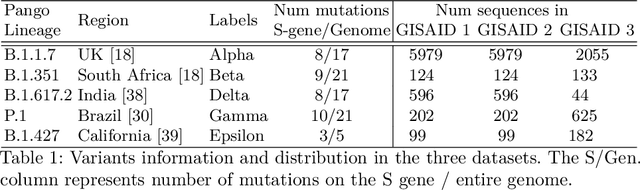
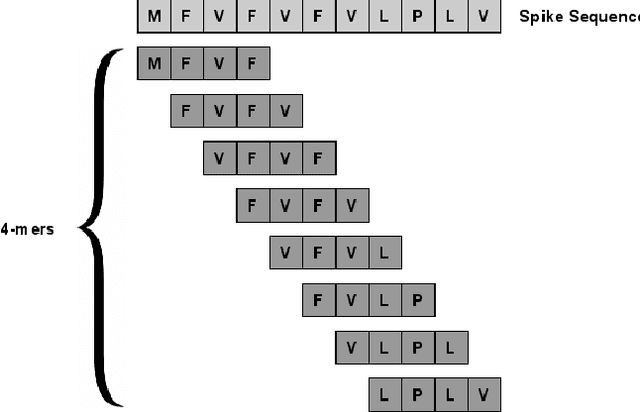
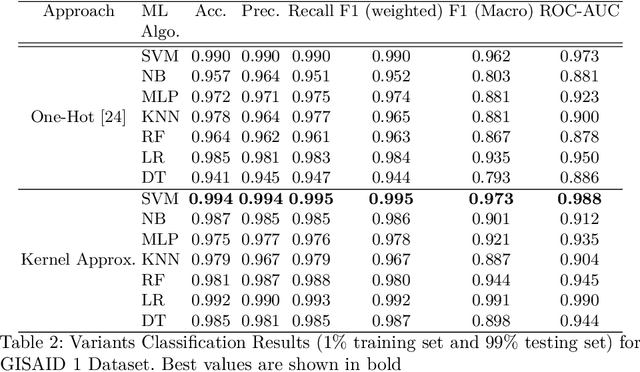
With the rapid spread of the novel coronavirus (COVID-19) across the globe and its continuous mutation, it is of pivotal importance to design a system to identify different known (and unknown) variants of SARS-CoV-2. Identifying particular variants helps to understand and model their spread patterns, design effective mitigation strategies, and prevent future outbreaks. It also plays a crucial role in studying the efficacy of known vaccines against each variant and modeling the likelihood of breakthrough infections. It is well known that the spike protein contains most of the information/variation pertaining to coronavirus variants. In this paper, we use spike sequences to classify different variants of the coronavirus in humans. We show that preserving the order of the amino acids helps the underlying classifiers to achieve better performance. We also show that we can train our model to outperform the baseline algorithms using only a small number of training samples ($1\%$ of the data). Finally, we show the importance of the different amino acids which play a key role in identifying variants and how they coincide with those reported by the USA's Centers for Disease Control and Prevention (CDC).
Effect of Analysis Window and Feature Selection on Classification of Hand Movements Using EMG Signal
Feb 13, 2020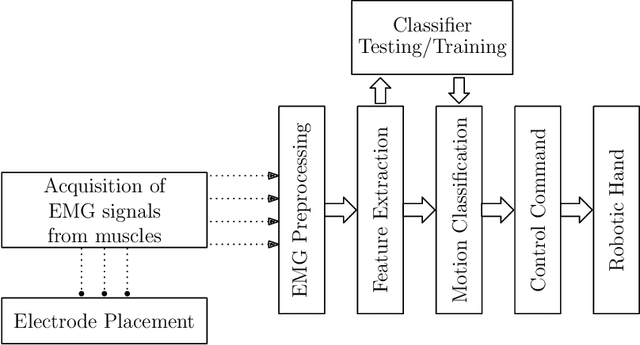
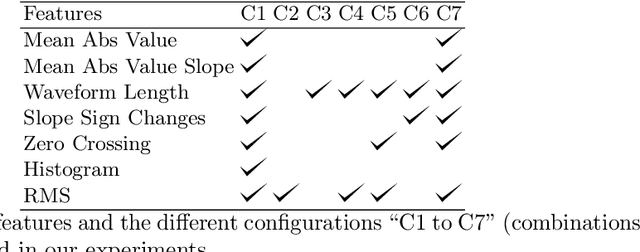
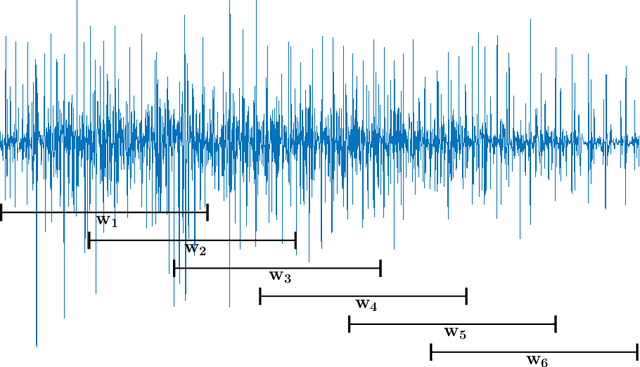

Electromyography (EMG) signals have been successfully employed for driving prosthetic limbs of a single or double degree of freedom. This principle works by using the amplitude of the EMG signals to decide between one or two simpler movements. This method underperforms as compare to the contemporary advances done at the mechanical, electronics, and robotics end, and it lacks intuition. Recently, research on myoelectric control based on pattern recognition (PR) shows promising results with the aid of machine learning classifiers. Using the approach termed as, EMG-PR, EMG signals are divided into analysis windows, and features are extracted for each window. These features are then fed to the machine learning classifiers as input. By offering multiple class movements and intuitive control, this method has the potential to power an amputated subject to perform everyday life movements. In this paper, we investigate the effect of the analysis window and feature selection on classification accuracy of different hand and wrist movements using time-domain features. We show that effective data preprocessing and optimum feature selection helps to improve the classification accuracy of hand movements. We use publicly available hand and wrist gesture dataset of $40$ intact subjects for experimentation. Results computed using different classification algorithms show that the proposed preprocessing and features selection outperforms the baseline and achieve up to $98\%$ classification accuracy.
Fair Allocation Based Soft Load Shedding
Feb 02, 2020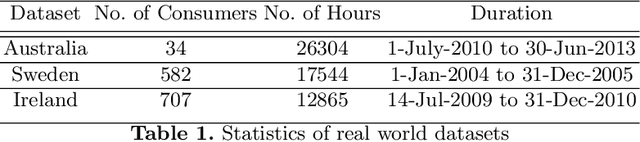
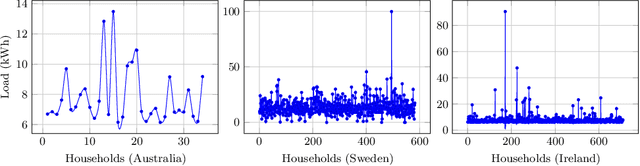
Renewable sources are taking center stage in electricity generation. Due to the intermittent nature of these renewable resources, the problem of the demand-supply gap arises. To solve this problem, several techniques have been proposed in the literature in terms of cost (adding peaker plants), availability of data (Demand Side Management "DSM"), hardware infrastructure (appliance controlling DSM) and safety (voltage reduction). However, these solutions are not fair in terms of electricity distribution. In many cases, although the available supply may not match the demand in peak hours, however, the total aggregated demand remains less than the total supply for the whole day. Load shedding (complete blackout) is a commonly used solution to deal with the demand-supply gap, which can cause substantial economic losses. To solve the demand-supply gap problem, we propose a solution called Soft Load Shedding (SLS), which assigns electricity quota to each household in a fair way. We measure the fairness of SLS by defining a function for household satisfaction level. We model the household utilities by parametric function and formulate the problem of SLS as a social welfare problem. We also consider revenue generated from the fair allocation as a performance measure. To evaluate our approach, extensive experiments have been performed on both synthetic and real-world datasets, and our model is compared with several baselines to show its effectiveness in terms of fair allocation and revenue generation.
Language Independent Sentiment Analysis
Jan 23, 2020
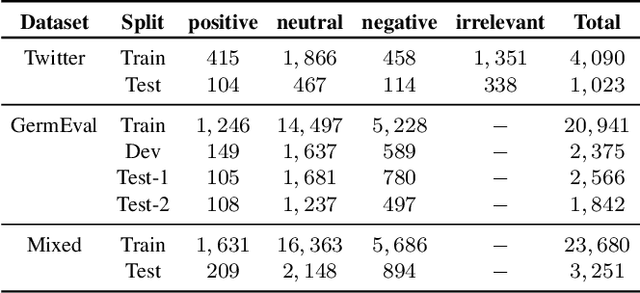

Social media platforms and online forums generate rapid and increasing amount of textual data. Businesses, government agencies, and media organizations seek to perform sentiment analysis on this rich text data. The results of these analytics are used for adapting marketing strategies, customizing products, security and various other decision makings. Sentiment analysis has been extensively studied and various methods have been developed for it with great success. These methods, however apply to texts written in a specific language. This limits applicability to a limited demographic and a specific geographic region. In this paper we propose a general approach for sentiment analysis on data containing texts from multiple languages. This enables all the applications to utilize the results of sentiment analysis in a language oblivious or language-independent fashion.
Hour-Ahead Load Forecasting Using AMI Data
Jan 08, 2020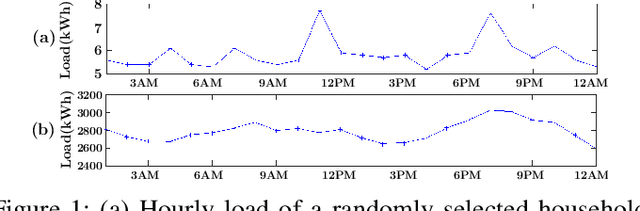
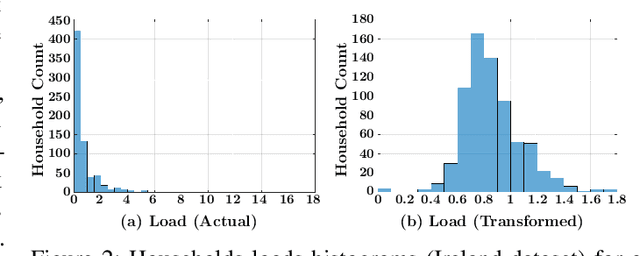

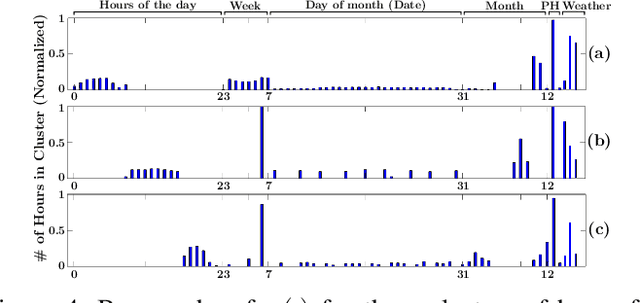
Accurate short-term load forecasting is essential for efficient operation of the power sector. Predicting load at a fine granularity such as individual households or buildings is challenging due to higher volatility and uncertainty in the load. In aggregate loads such as at grids level, the inherent stochasticity and fluctuations are averaged-out, the problem becomes substantially easier. We propose an approach for short-term load forecasting at individual consumers (households) level, called Forecasting using Matrix Factorization (FMF). FMF does not use any consumers' demographic or activity patterns information. Therefore, it can be applied to any locality with the readily available smart meters and weather data. We perform extensive experiments on three benchmark datasets and demonstrate that FMF significantly outperforms the computationally expensive state-of-the-art methods for this problem. We achieve up to 26.5% and 24.4 % improvement in RMSE over Regression Tree and Support Vector Machine, respectively and up to 36% and 73.2% improvement in MAPE over Random Forest and Long Short-Term Memory neural network, respectively.