 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEpiFormer: Learning Antigen-Antibody Interactions for Epitope Prediction via Geometric Deep Learning
Jun 02, 2026Antibodies neutralize foreign antigens by binding to specific surface regions called epitopes. Computational epitope prediction is critical for understanding immune recognition and guiding antibody engineering. However, existing methods face three fundamental challenges: antibody-aware models encode each chain independently and combine them only at a late stage, failing to capture co-dependent structural features that define binding interfaces, whereas severe class imbalance and scarcity of known antibody-antigen complexes render standard training objectives ineffective. We propose EpiFormer, a general encoder-decoder framework that addresses these challenges jointly. Our key design principle is interleaved cross-attention within GNN encoding layers, enabling bidirectional antigen-antibody information flow throughout representation learning rather than only at the output. This early-fusion principle is backbone-agnostic, providing consistent gains across GNN architectures from simple GCNs to equivariant models. We further show that sparsity-aware objectives are effective when paired with early-fusion architectures for the epitope prediction task. EpiFormer improves over the previous best method by over 40% in F1 score on standard benchmarks, demonstrating generalizability and cross-dataset transferability. Notably, EpiFormer discovers known biological principles as emergent behaviors of end-to-end training, where the learned cross-attention gates favor antigen-to-antibody information flow, consistent with the asymmetric roles of the two chains at the binding interface, and the model's preference for geometric over evolutionary features aligns with the established finding that epitope residues are not evolutionarily conserved. The source code is available at: https://github.com/mansoor181/epiformer.git
ConTact: Contact-First Antibody CDR Design via Explicit Interface Reasoning
May 20, 2026Computational antibody CDR design methods condition on antigen structure to generate binding loops, yet existing architectures conflate two fundamentally distinct sub-problems: identifying which CDR positions will contact the antigen, and selecting amino acids at those positions. This conflation forces models to learn contact reasoning implicitly through uniform message passing, diluting antigen signal across all positions equally. We introduce ConTact, a contact-then-act architecture that explicitly decomposes CDR design into three cascaded stages: learning surface complementarity fingerprints, predicting CDR-antigen contacts, and injecting contact-gated antigen features into the sequence head. A distance-biased cross-attention module encodes geometric priors favoring spatial neighbors, while a contact-weighted cross-entropy loss concentrates gradient signal on binding-critical positions. On CHIMERA-Bench dataset, ConTact achieves the best structural quality (7% RMSD improvement over the next-best baseline), best epitope awareness (10% F1 score over GNN baselines), and competitive sequence recovery (AAR 0.38) among several CDR-H3 design baselines.
EvoStruct: Bridging Evolutionary and Structural Priors for Antibody CDR Design via Protein Language Model Adaptation
May 20, 2026Equivariant graph neural network (GNN) methods for antibody complementarity-determining region (CDR) design achieve the highest sequence recovery but suffer from severe vocabulary collapse. The current best GNN methods over-predict very few amino acids, such as tyrosine and glycine, while ignoring functionally important residues. We trace this failure to GNN encoders learning amino acid distributions de novo from limited structural data, discarding substitution patterns encoded in evolutionary databases. To resolve this, we propose EvoStruct, which bridges a frozen protein language model (PLM) with 3D structural context from an E(3)-equivariant GNN via a cross-attention adapter. Unlike prior PLM-structure adapters for general protein design, EvoStruct targets the vocabulary collapse problem specific to CDR design through progressive PLM unfreezing and R-Drop consistency regularization. On the CHIMERA-Bench dataset, EvoStruct achieves the highest amino acid recovery and lowest perplexity among several antibody design methods, improving sequence recovery by 16% and reducing perplexity by 43% relative to the best GNN baselines, while recovering 2.3x greater amino acid diversity and the highest binding-pair correlation with ground truth.
AgForce Enables Antigen-conditioned Generative Antibody Design
May 20, 2026Antibody design methods condition on antigen structure to generate complementarity-determining regions (CDR), yet a systematic evaluation of baseline methods reveals that they largely ignore the antigen input. We identify three failure modes that explain this behavior. Antigen blindness arises because models derive predictions from antibody framework context rather than antigen information, producing nearly identical CDRs regardless of the target. Vocabulary collapse reduces predicted amino acids to three to five per position, far below the ground truth distribution in native sequences. Moreover, any model trained with standard per-position cross-entropy converges to the positional marginal distribution, making it provably unable to produce antigen-specific sequence predictions. We propose a novel encoder-decoder architecture called AgForce, that uses a graph neural network (GNN) as the encoder and specialized decoders for sequence-structure co-design. Specifically, we apply framework dropout, gated bottlenecks, and hyperbolic cross attention that prevent the antibody shortcut path. In the decoder, a Mixture Density Network (MDN) sequence head with Potts-like pairwise coupling and annealed Multiple Choice Learning (aMCL) replaces the cross-entropy objective with a multi-component distribution whose optimal solution differs from the positional marginal. An antigen cycle consistency head routes gradients through the sequence decoder, forcing predicted distributions to encode antigen identity. AgForce achieves the best binding quality and sequence recovery simultaneously on the CHIMERA-Bench dataset, improving amino acid recovery by 8% over the strongest sequence baseline while surpassing the baselines across all interface metrics, and nearly doubling the effective vocabulary of GNN methods. The source code is available at: https://github.com/mansoor181/ag-force.git
CHIMERA-Bench: A Benchmark Dataset for Epitope-Specific Antibody Design
Mar 13, 2026Computational antibody design has seen rapid methodological progress, with dozens of deep generative methods proposed in the past three years, yet the field lacks a standardized benchmark for fair comparison and model development. These methods are evaluated on different SAbDab snapshots, non-overlapping test sets, and incompatible metrics, and the literature fragments the design problem into numerous sub-tasks with no common definition. We introduce \textsc{Chimera-Bench} (\textbf{C}DR \textbf{M}odeling with \textbf{E}pitope-guided \textbf{R}edesign), a unified benchmark built around a single canonical task: \emph{epitope-conditioned CDR sequence-structure co-design}. \textsc{Chimera-Bench} provides (1) a curated, deduplicated dataset of \textbf{2,922} antibody-antigen complexes with epitope and paratope annotations; (2) three biologically motivated splits testing generalization to unseen epitopes, unseen antigen folds, and prospective temporal targets; and (3) a comprehensive evaluation protocol with five metric groups including novel epitope-specificity measures. We benchmark representative methods spanning different generative paradigms and report results across all splits. \textsc{Chimera-Bench} is the largest dataset of its kind for the antibody design problem, allowing the community to develop and test novel methods and evaluate their generalizability. The source code and data are available at: https://github.com/mansoor181/chimera-bench.git
Boosting t-SNE Efficiency for Sequencing Data: Insights from Kernel Selection
Dec 17, 2025



Dimensionality reduction techniques are essential for visualizing and analyzing high-dimensional biological sequencing data. t-distributed Stochastic Neighbor Embedding (t-SNE) is widely used for this purpose, traditionally employing the Gaussian kernel to compute pairwise similarities. However, the Gaussian kernel's lack of data-dependence and computational overhead limit its scalability and effectiveness for categorical biological sequences. Recent work proposed the isolation kernel as an alternative, yet it may not optimally capture sequence similarities. In this study, we comprehensively evaluate nine different kernel functions for t-SNE applied to molecular sequences, using three embedding methods: One-Hot Encoding, Spike2Vec, and minimizers. Through both subjective visualization and objective metrics (including neighborhood preservation scores), we demonstrate that the cosine similarity kernel in general outperforms other kernels, including Gaussian and isolation kernels, achieving superior runtime efficiency and better preservation of pairwise distances in low-dimensional space. We further validate our findings through extensive classification and clustering experiments across six diverse biological datasets (Spike7k, Host, ShortRead, Rabies, Genome, and Breast Cancer), employing multiple machine learning algorithms and evaluation metrics. Our results show that kernel selection significantly impacts not only visualization quality but also downstream analytical tasks, with the cosine similarity kernel providing the most robust performance across different data types and embedding strategies, making it particularly suitable for large-scale biological sequence analysis.
EfficientNet in Digital Twin-based Cardiac Arrest Prediction and Analysis
Sep 09, 2025Cardiac arrest is one of the biggest global health problems, and early identification and management are key to enhancing the patient's prognosis. In this paper, we propose a novel framework that combines an EfficientNet-based deep learning model with a digital twin system to improve the early detection and analysis of cardiac arrest. We use compound scaling and EfficientNet to learn the features of cardiovascular images. In parallel, the digital twin creates a realistic and individualized cardiovascular system model of the patient based on data received from the Internet of Things (IoT) devices attached to the patient, which can help in the constant assessment of the patient and the impact of possible treatment plans. As shown by our experiments, the proposed system is highly accurate in its prediction abilities and, at the same time, efficient. Combining highly advanced techniques such as deep learning and digital twin (DT) technology presents the possibility of using an active and individual approach to predicting cardiac disease.
Enhancing Privacy Preservation and Reducing Analysis Time with Federated Transfer Learning in Digital Twins-based Computed Tomography Scan Analysis
Sep 09, 2025The application of Digital Twin (DT) technology and Federated Learning (FL) has great potential to change the field of biomedical image analysis, particularly for Computed Tomography (CT) scans. This paper presents Federated Transfer Learning (FTL) as a new Digital Twin-based CT scan analysis paradigm. FTL uses pre-trained models and knowledge transfer between peer nodes to solve problems such as data privacy, limited computing resources, and data heterogeneity. The proposed framework allows real-time collaboration between cloud servers and Digital Twin-enabled CT scanners while protecting patient identity. We apply the FTL method to a heterogeneous CT scan dataset and assess model performance using convergence time, model accuracy, precision, recall, F1 score, and confusion matrix. It has been shown to perform better than conventional FL and Clustered Federated Learning (CFL) methods with better precision, accuracy, recall, and F1-score. The technique is beneficial in settings where the data is not independently and identically distributed (non-IID), and it offers reliable, efficient, and secure solutions for medical diagnosis. These findings highlight the possibility of using FTL to improve decision-making in digital twin-based CT scan analysis, secure and efficient medical image analysis, promote privacy, and open new possibilities for applying precision medicine and smart healthcare systems.
Sequence Analysis Using the Bezier Curve
Mar 18, 2025



The analysis of sequences (e.g., protein, DNA, and SMILES string) is essential for disease diagnosis, biomaterial engineering, genetic engineering, and drug discovery domains. Conventional analytical methods focus on transforming sequences into numerical representations for applying machine learning/deep learning-based sequence characterization. However, their efficacy is constrained by the intrinsic nature of deep learning (DL) models, which tend to exhibit suboptimal performance when applied to tabular data. An alternative group of methodologies endeavors to convert biological sequences into image forms by applying the concept of Chaos Game Representation (CGR). However, a noteworthy drawback of these methods lies in their tendency to map individual elements of the sequence onto a relatively small subset of designated pixels within the generated image. The resulting sparse image representation may not adequately encapsulate the comprehensive sequence information, potentially resulting in suboptimal predictions. In this study, we introduce a novel approach to transform sequences into images using the B\'ezier curve concept for element mapping. Mapping the elements onto a curve enhances the sequence information representation in the respective images, hence yielding better DL-based classification performance. We employed different sequence datasets to validate our system by using different classification tasks, and the results illustrate that our B\'ezier curve method is able to achieve good performance for all the tasks.
Hilbert Curve Based Molecular Sequence Analysis
Dec 29, 2024
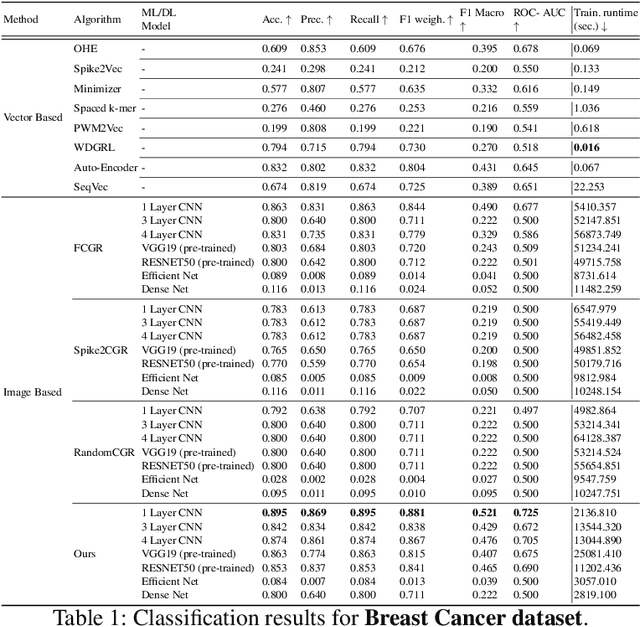

Accurate molecular sequence analysis is a key task in the field of bioinformatics. To apply molecular sequence classification algorithms, we first need to generate the appropriate representations of the sequences. Traditional numeric sequence representation techniques are mostly based on sequence alignment that faces limitations in the form of lack of accuracy. Although several alignment-free techniques have also been introduced, their tabular data form results in low performance when used with Deep Learning (DL) models compared to the competitive performance observed in the case of image-based data. To find a solution to this problem and to make Deep Learning (DL) models function to their maximum potential while capturing the important spatial information in the sequence data, we propose a universal Hibert curve-based Chaos Game Representation (CGR) method. This method is a transformative function that involves a novel Alphabetic index mapping technique used in constructing Hilbert curve-based image representation from molecular sequences. Our method can be globally applied to any type of molecular sequence data. The Hilbert curve-based image representations can be used as input to sophisticated vision DL models for sequence classification. The proposed method shows promising results as it outperforms current state-of-the-art methods by achieving a high accuracy of $94.5$\% and an F1 score of $93.9\%$ when tested with the CNN model on the lung cancer dataset. This approach opens up a new horizon for exploring molecular sequence analysis using image classification methods.