 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssayBench: An Assay-Level Virtual Cell Benchmark for LLMs and Agents
May 11, 2026Recent advances in machine learning and large-scale biological data collections have revived the prospect of building a virtual cell, a computational model of cellular behavior that could accelerate biological discovery. One of the most compelling promises of this vision is the ability to perform in silico phenotypic screens, in which a model predicts the effects of cellular perturbations in unseen biological contexts. This task combines heterogeneous textual inputs with diverse phenotypic outputs, making it particularly well-suited to LLMs and agentic systems. Yet, no standard benchmark currently exists for this task, as existing efforts focus on narrower molecular readouts that are only indirectly aligned with the phenotypic endpoints driving many real-world drug discovery workflows. In this work, we present AssayBench, a benchmark for phenotypic screen prediction, built from 1,920 publicly available CRISPR screens spanning five broad classes of cellular phenotypes. We formulate the screen prediction task as a gene rank prediction for each screen and introduce the adjusted nDCG, a continuous metric for comparing performance across heterogeneous assays. Our extensive evaluation shows that existing methods remain far from empirically estimated performance ceilings and zero-shot generalist LLMs outperform biology-specific LLMs and trainable baselines. Optimization techniques such as fine-tuning, ensembling, and prompt optimization can further improve LLM performance on this task. Overall, AssayBench offers a practical testbed for measuring progress toward in silico phenotypic screening and, more broadly, virtual cell models.
BenchGuard: Who Guards the Benchmarks? Automated Auditing of LLM Agent Benchmarks
Apr 27, 2026As benchmarks grow in complexity, many apparent agent failures are not failures of the agent at all - they are failures of the benchmark itself: broken specifications, implicit assumptions, and rigid evaluation scripts that penalize valid alternative approaches. We propose employing frontier LLMs as systematic auditors of evaluation infrastructure, and realize this vision through BenchGuard, the first automated auditing framework for task-oriented, execution-based agent benchmarks. BenchGuard cross-verifies all benchmark artifacts via structured LLM protocols, optionally incorporating agent solutions or execution traces as additional diagnostic evidence. Deployed on two prominent scientific benchmarks, BenchGuard identified 12 author-confirmed issues in ScienceAgentBench - including fatal errors rendering tasks unsolvable - and exactly matched 83.3% of expert-identified issues on the BIXBench Verified-50 subset, catching defects that prior human review missed entirely. A full audit of 50 complex bioinformatics tasks costs under USD 15, making automated benchmark auditing a practical and valuable complement to human review. These findings point toward AI-assisted benchmark development, where frontier models serve not only as subjects of evaluation but as active participants in validating the evaluation infrastructure itself.
Modeling trajectories of mental health: challenges and opportunities
Dec 04, 2016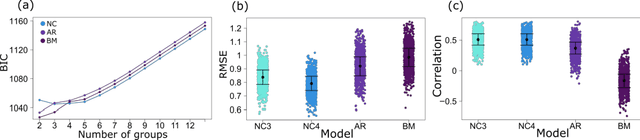
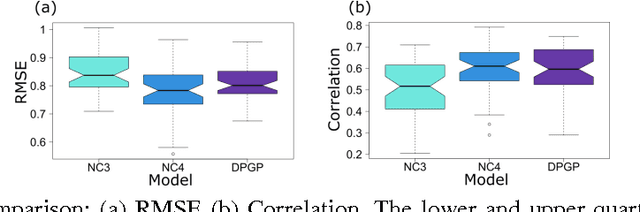
More than two thirds of mental health problems have their onset during childhood or adolescence. Identifying children at risk for mental illness later in life and predicting the type of illness is not easy. We set out to develop a platform to define subtypes of childhood social-emotional development using longitudinal, multifactorial trait-based measures. Subtypes discovered through this study could ultimately advance psychiatric knowledge of the early behavioural signs of mental illness. To this extent we have examined two types of models: latent class mixture models and GP-based models. Our findings indicate that while GP models come close in accuracy of predicting future trajectories, LCMMs predict the trajectories as well in a fraction of the time. Unfortunately, neither of the models are currently accurate enough to lead to immediate clinical impact. The available data related to the development of childhood mental health is often sparse with only a few time points measured and require novel methods with improved efficiency and accuracy.
Using the Gene Ontology Hierarchy when Predicting Gene Function
May 09, 2012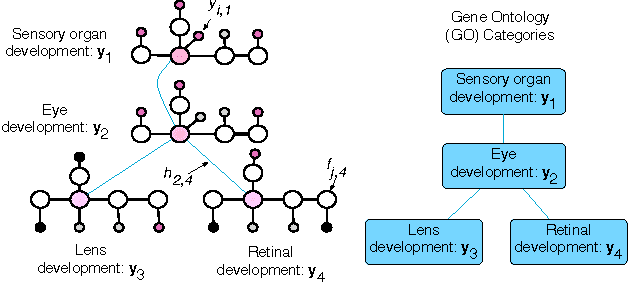
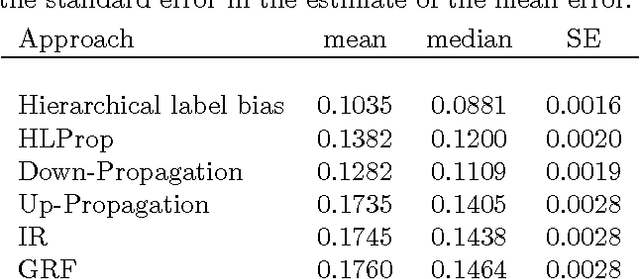
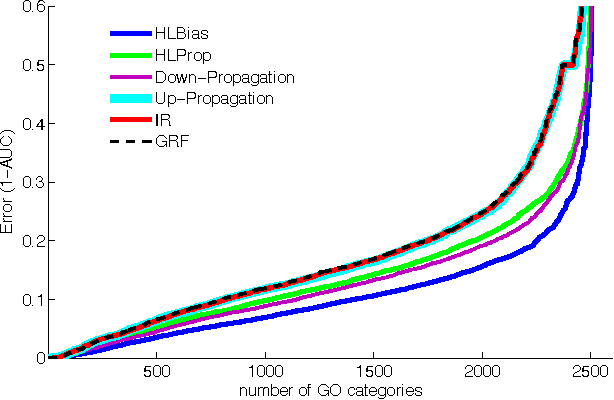
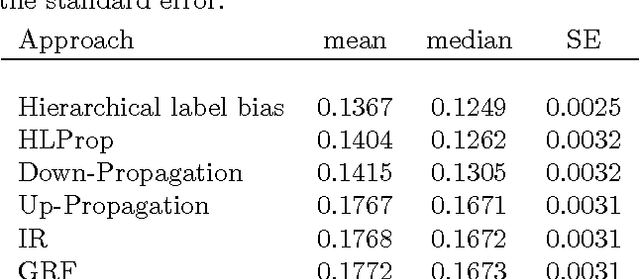
The problem of multilabel classification when the labels are related through a hierarchical categorization scheme occurs in many application domains such as computational biology. For example, this problem arises naturally when trying to automatically assign gene function using a controlled vocabularies like Gene Ontology. However, most existing approaches for predicting gene functions solve independent classification problems to predict genes that are involved in a given function category, independently of the rest. Here, we propose two simple methods for incorporating information about the hierarchical nature of the categorization scheme. In the first method, we use information about a gene's previous annotation to set an initial prior on its label. In a second approach, we extend a graph-based semi-supervised learning algorithm for predicting gene function in a hierarchy. We show that we can efficiently solve this problem by solving a linear system of equations. We compare these approaches with a previous label reconciliation-based approach. Results show that using the hierarchy information directly, compared to using reconciliation methods, improves gene function prediction.