 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssayBench: An Assay-Level Virtual Cell Benchmark for LLMs and Agents
May 11, 2026Recent advances in machine learning and large-scale biological data collections have revived the prospect of building a virtual cell, a computational model of cellular behavior that could accelerate biological discovery. One of the most compelling promises of this vision is the ability to perform in silico phenotypic screens, in which a model predicts the effects of cellular perturbations in unseen biological contexts. This task combines heterogeneous textual inputs with diverse phenotypic outputs, making it particularly well-suited to LLMs and agentic systems. Yet, no standard benchmark currently exists for this task, as existing efforts focus on narrower molecular readouts that are only indirectly aligned with the phenotypic endpoints driving many real-world drug discovery workflows. In this work, we present AssayBench, a benchmark for phenotypic screen prediction, built from 1,920 publicly available CRISPR screens spanning five broad classes of cellular phenotypes. We formulate the screen prediction task as a gene rank prediction for each screen and introduce the adjusted nDCG, a continuous metric for comparing performance across heterogeneous assays. Our extensive evaluation shows that existing methods remain far from empirically estimated performance ceilings and zero-shot generalist LLMs outperform biology-specific LLMs and trainable baselines. Optimization techniques such as fine-tuning, ensembling, and prompt optimization can further improve LLM performance on this task. Overall, AssayBench offers a practical testbed for measuring progress toward in silico phenotypic screening and, more broadly, virtual cell models.
CloudPred: Predicting Patient Phenotypes From Single-cell RNA-seq
Oct 13, 2021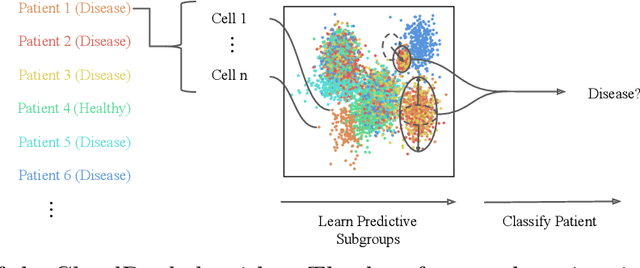
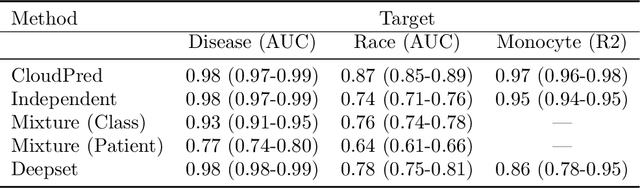
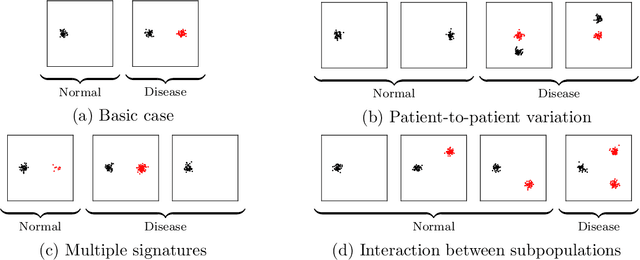
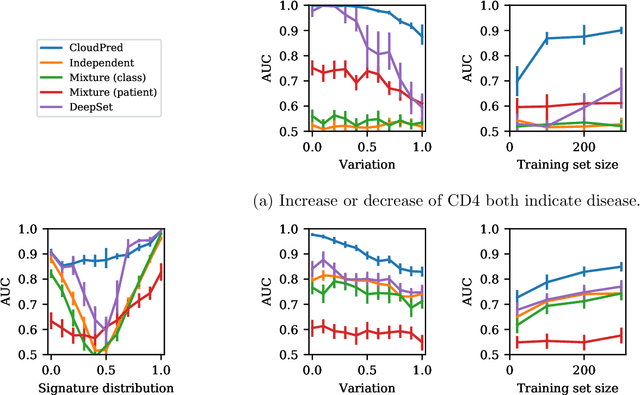
Single-cell RNA sequencing (scRNA-seq) has the potential to provide powerful, high-resolution signatures to inform disease prognosis and precision medicine. This paper takes an important first step towards this goal by developing an interpretable machine learning algorithm, CloudPred, to predict individuals' disease phenotypes from their scRNA-seq data. Predicting phenotype from scRNA-seq is challenging for standard machine learning methods -- the number of cells measured can vary by orders of magnitude across individuals and the cell populations are also highly heterogeneous. Typical analysis creates pseudo-bulk samples which are biased toward prior annotations and also lose the single cell resolution. CloudPred addresses these challenges via a novel end-to-end differentiable learning algorithm which is coupled with a biologically informed mixture of cell types model. CloudPred automatically infers the cell subpopulation that are salient for the phenotype without prior annotations. We developed a systematic simulation platform to evaluate the performance of CloudPred and several alternative methods we propose, and find that CloudPred outperforms the alternative methods across several settings. We further validated CloudPred on a real scRNA-seq dataset of 142 lupus patients and controls. CloudPred achieves AUROC of 0.98 while identifying a specific subpopulation of CD4 T cells whose presence is highly indicative of lupus. CloudPred is a powerful new framework to predict clinical phenotypes from scRNA-seq data and to identify relevant cells.