 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSamuel S. Schoenholz
Intriguing Properties of Adversarial Examples
Nov 08, 2017
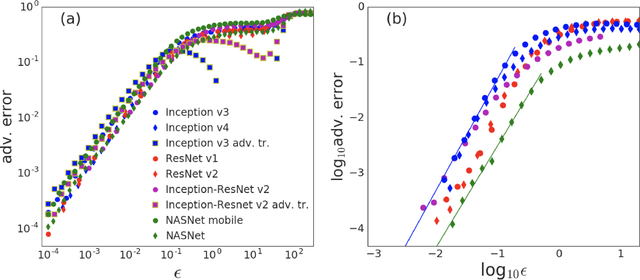

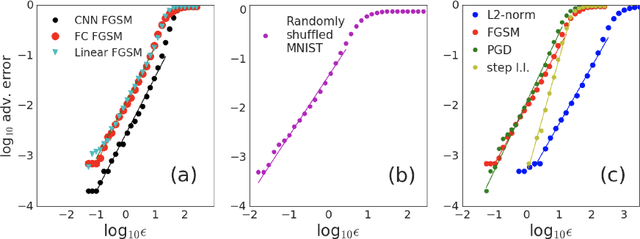
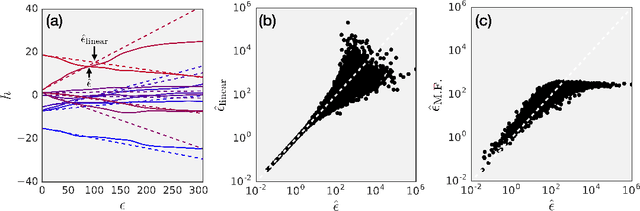
It is becoming increasingly clear that many machine learning classifiers are vulnerable to adversarial examples. In attempting to explain the origin of adversarial examples, previous studies have typically focused on the fact that neural networks operate on high dimensional data, they overfit, or they are too linear. Here we argue that the origin of adversarial examples is primarily due to an inherent uncertainty that neural networks have about their predictions. We show that the functional form of this uncertainty is independent of architecture, dataset, and training protocol; and depends only on the statistics of the logit differences of the network, which do not change significantly during training. This leads to adversarial error having a universal scaling, as a power-law, with respect to the size of the adversarial perturbation. We show that this universality holds for a broad range of datasets (MNIST, CIFAR10, ImageNet, and random data), models (including state-of-the-art deep networks, linear models, adversarially trained networks, and networks trained on randomly shuffled labels), and attacks (FGSM, step l.l., PGD). Motivated by these results, we study the effects of reducing prediction entropy on adversarial robustness. Finally, we study the effect of network architectures on adversarial sensitivity. To do this, we use neural architecture search with reinforcement learning to find adversarially robust architectures on CIFAR10. Our resulting architecture is more robust to white \emph{and} black box attacks compared to previous attempts.
A Correspondence Between Random Neural Networks and Statistical Field Theory
Oct 18, 2017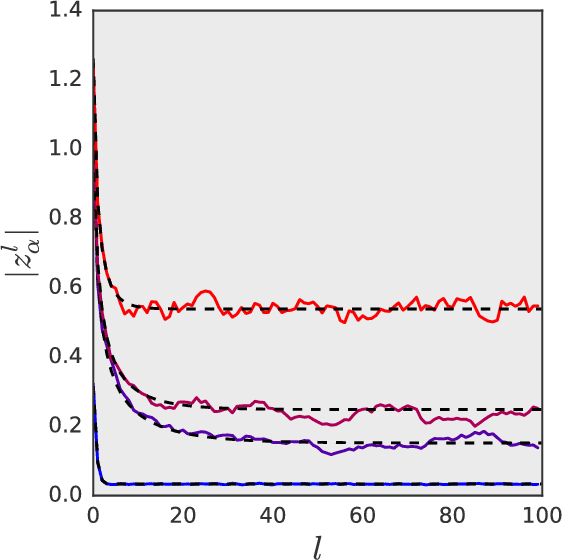
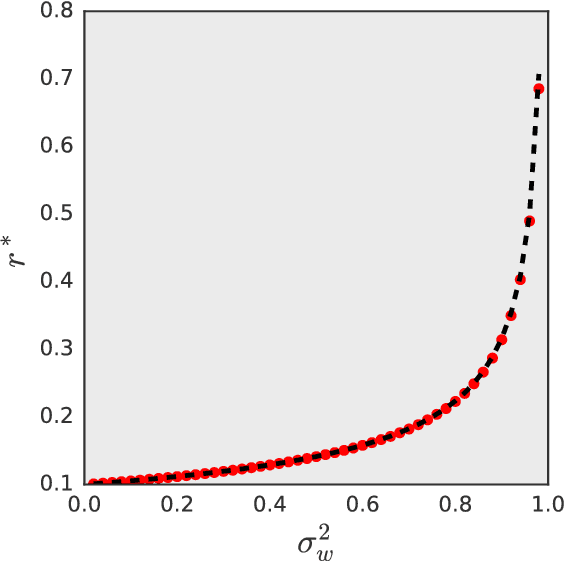
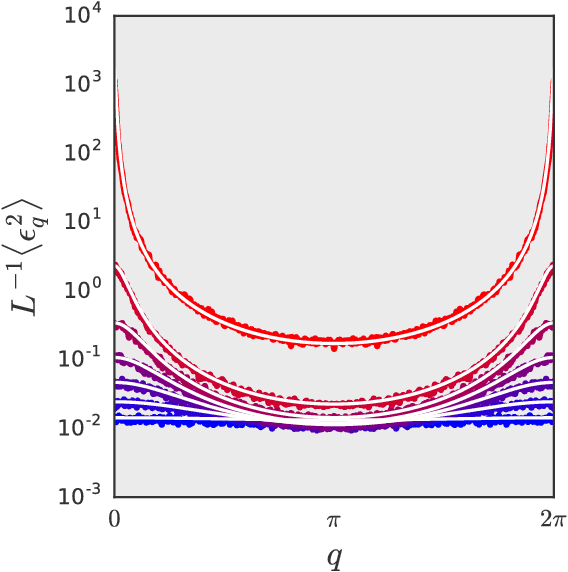
A number of recent papers have provided evidence that practical design questions about neural networks may be tackled theoretically by studying the behavior of random networks. However, until now the tools available for analyzing random neural networks have been relatively ad-hoc. In this work, we show that the distribution of pre-activations in random neural networks can be exactly mapped onto lattice models in statistical physics. We argue that several previous investigations of stochastic networks actually studied a particular factorial approximation to the full lattice model. For random linear networks and random rectified linear networks we show that the corresponding lattice models in the wide network limit may be systematically approximated by a Gaussian distribution with covariance between the layers of the network. In each case, the approximate distribution can be diagonalized by Fourier transformation. We show that this approximation accurately describes the results of numerical simulations of wide random neural networks. Finally, we demonstrate that in each case the large scale behavior of the random networks can be approximated by an effective field theory.
Combining Machine Learning and Physics to Understand Glassy Systems
Sep 23, 2017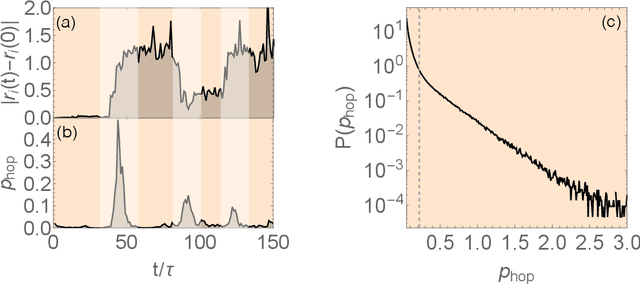
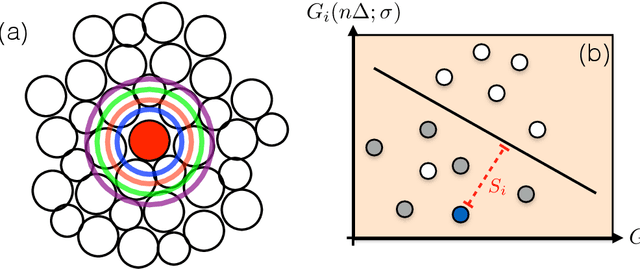
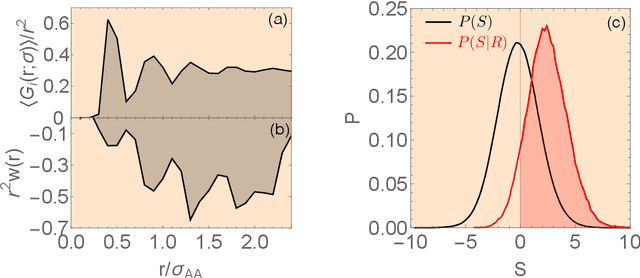
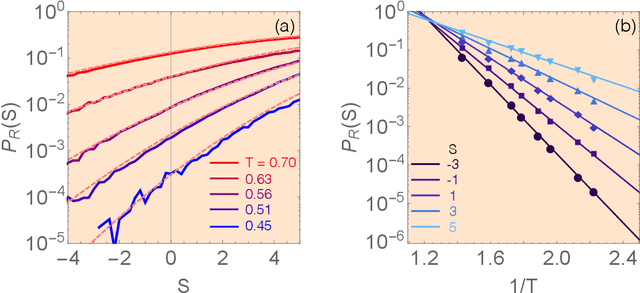
Our understanding of supercooled liquids and glasses has lagged significantly behind that of simple liquids and crystalline solids. This is in part due to the many possibly relevant degrees of freedom that are present due to the disorder inherent to these systems and in part to non-equilibrium effects which are difficult to treat in the standard context of statistical physics. Together these issues have resulted in a field whose theories are under-constrained by experiment and where fundamental questions are still unresolved. Mean field results have been successful in infinite dimensions but it is unclear to what extent they apply to realistic systems and assume uniform local structure. At odds with this are theories premised on the existence of structural defects. However, until recently it has been impossible to find structural signatures that are predictive of dynamics. Here we summarize and recast the results from several recent papers offering a data driven approach to building a phenomenological theory of disordered materials by combining machine learning with physical intuition.
Neural Message Passing for Quantum Chemistry
Jun 12, 2017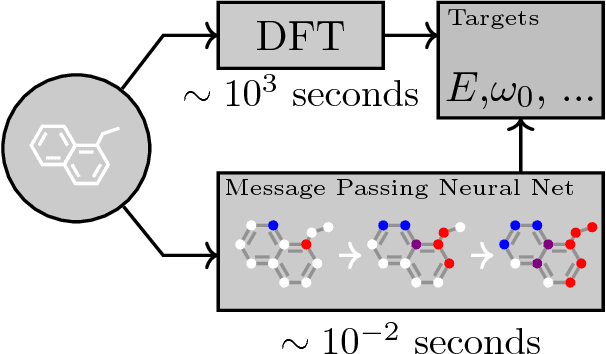

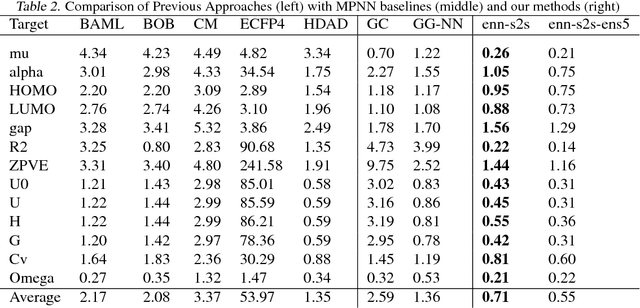
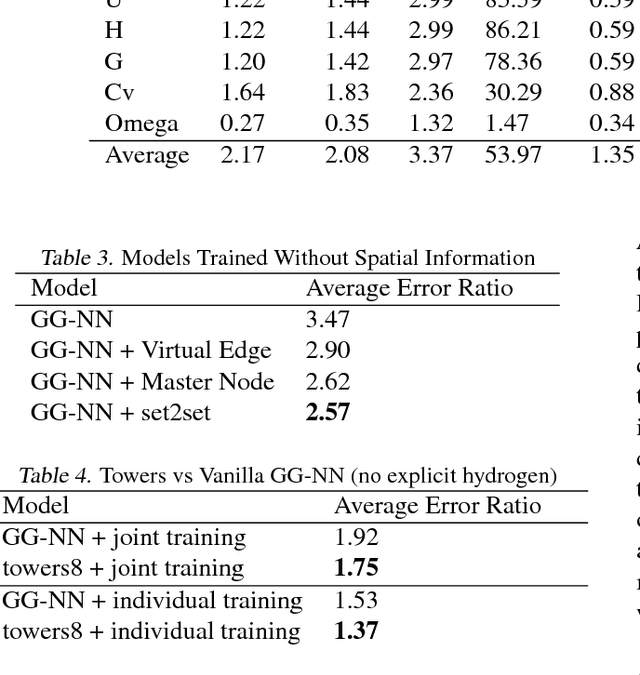
Supervised learning on molecules has incredible potential to be useful in chemistry, drug discovery, and materials science. Luckily, several promising and closely related neural network models invariant to molecular symmetries have already been described in the literature. These models learn a message passing algorithm and aggregation procedure to compute a function of their entire input graph. At this point, the next step is to find a particularly effective variant of this general approach and apply it to chemical prediction benchmarks until we either solve them or reach the limits of the approach. In this paper, we reformulate existing models into a single common framework we call Message Passing Neural Networks (MPNNs) and explore additional novel variations within this framework. Using MPNNs we demonstrate state of the art results on an important molecular property prediction benchmark; these results are strong enough that we believe future work should focus on datasets with larger molecules or more accurate ground truth labels.
Deep Information Propagation
Apr 04, 2017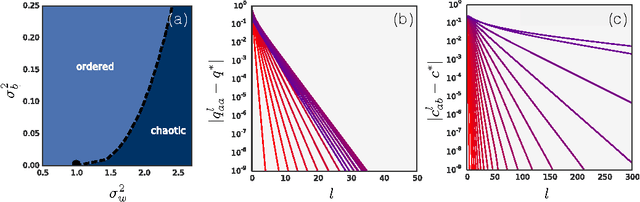
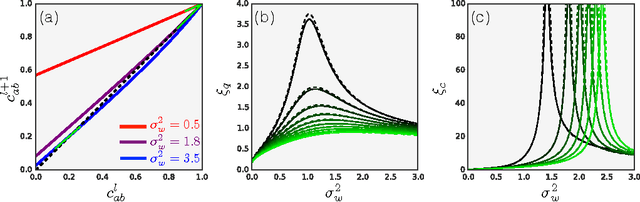
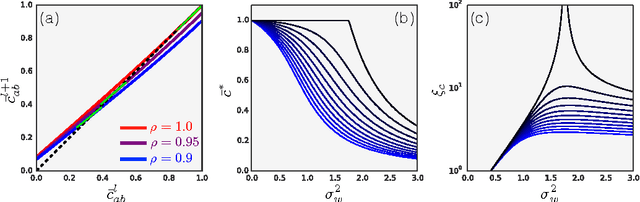
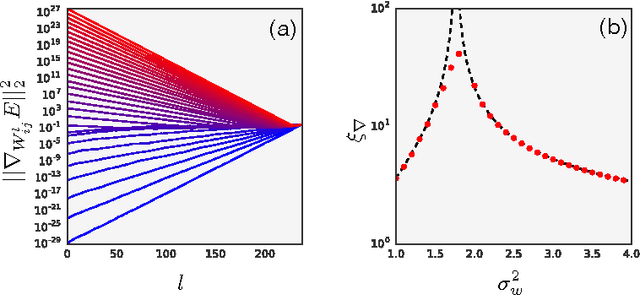
We study the behavior of untrained neural networks whose weights and biases are randomly distributed using mean field theory. We show the existence of depth scales that naturally limit the maximum depth of signal propagation through these random networks. Our main practical result is to show that random networks may be trained precisely when information can travel through them. Thus, the depth scales that we identify provide bounds on how deep a network may be trained for a specific choice of hyperparameters. As a corollary to this, we argue that in networks at the edge of chaos, one of these depth scales diverges. Thus arbitrarily deep networks may be trained only sufficiently close to criticality. We show that the presence of dropout destroys the order-to-chaos critical point and therefore strongly limits the maximum trainable depth for random networks. Finally, we develop a mean field theory for backpropagation and we show that the ordered and chaotic phases correspond to regions of vanishing and exploding gradient respectively.