 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeuroRule: A Connectionist Approach to Data Mining
Jan 05, 2017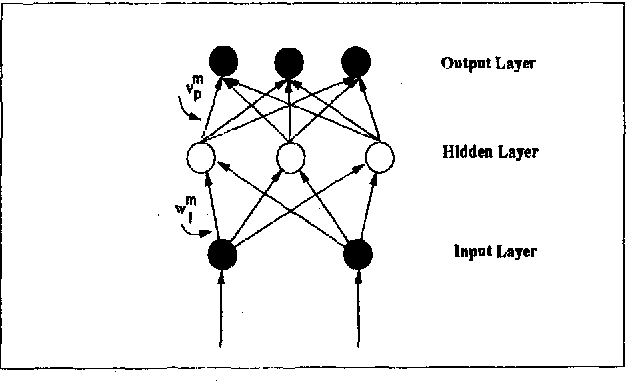


Classification, which involves finding rules that partition a given data set into disjoint groups, is one class of data mining problems. Approaches proposed so far for mining classification rules for large databases are mainly decision tree based symbolic learning methods. The connectionist approach based on neural networks has been thought not well suited for data mining. One of the major reasons cited is that knowledge generated by neural networks is not explicitly represented in the form of rules suitable for verification or interpretation by humans. This paper examines this issue. With our newly developed algorithms, rules which are similar to, or more concise than those generated by the symbolic methods can be extracted from the neural networks. The data mining process using neural networks with the emphasis on rule extraction is described. Experimental results and comparison with previously published works are presented.
Gene selection for cancer classification using a hybrid of univariate and multivariate feature selection methods
Jun 05, 2015
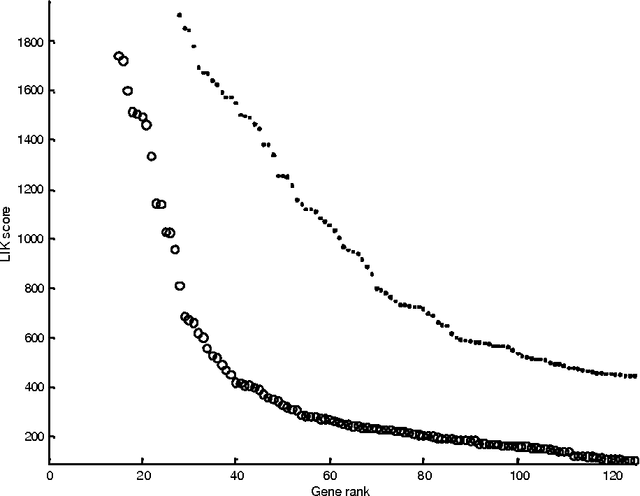

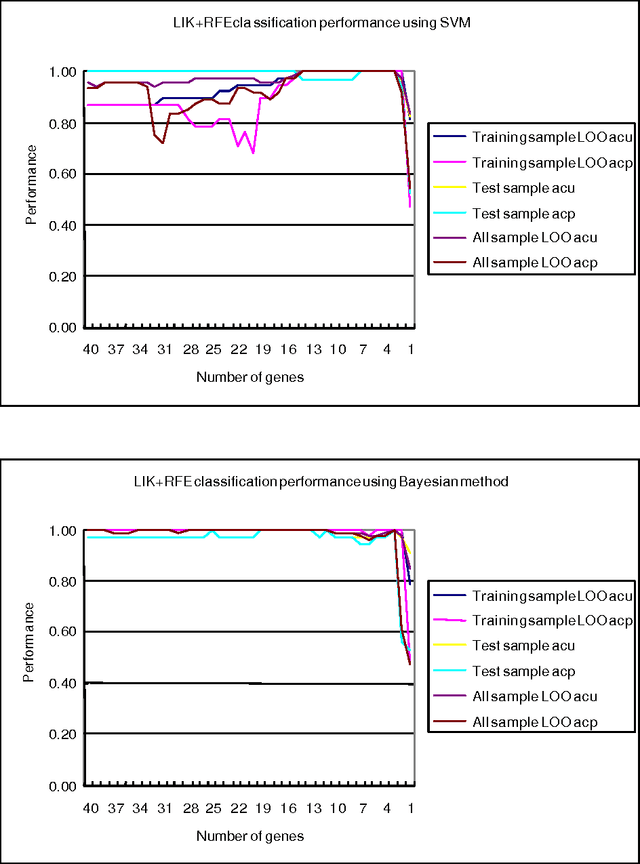
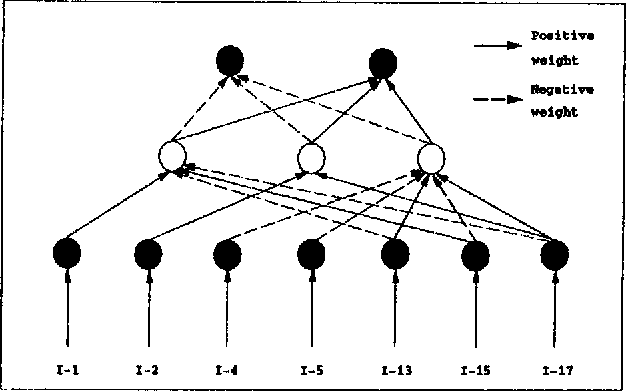
Various approaches to gene selection for cancer classification based on microarray data can be found in the literature and they may be grouped into two categories: univariate methods and multivariate methods. Univariate methods look at each gene in the data in isolation from others. They measure the contribution of a particular gene to the classification without considering the presence of the other genes. In contrast, multivariate methods measure the relative contribution of a gene to the classification by taking the other genes in the data into consideration. Multivariate methods select fewer genes in general. However, the selection process of multivariate methods may be sensitive to the presence of irrelevant genes, noises in the expression and outliers in the training data. At the same time, the computational cost of multivariate methods is high. To overcome the disadvantages of the two types of approaches, we propose a hybrid method to obtain gene sets that are small and highly discriminative. We devise our hybrid method from the univariate Maximum Likelihood method (LIK) and the multivariate Recursive Feature Elimination method (RFE). We analyze the properties of these methods and systematically test the effectiveness of our proposed method on two cancer microarray datasets. Our experiments on a leukemia dataset and a small, round blue cell tumors dataset demonstrate the effectiveness of our hybrid method. It is able to discover sets consisting of fewer genes than those reported in the literature and at the same time achieve the same or better prediction accuracy.