 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeActive Learning of Uniformly Accurate Inter-atomic Potentials for Materials Simulation
Oct 28, 2018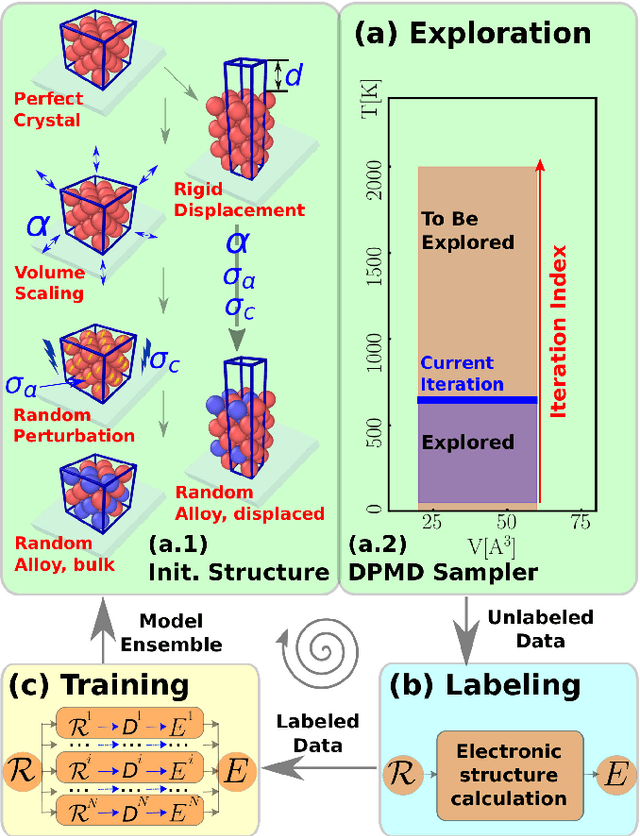
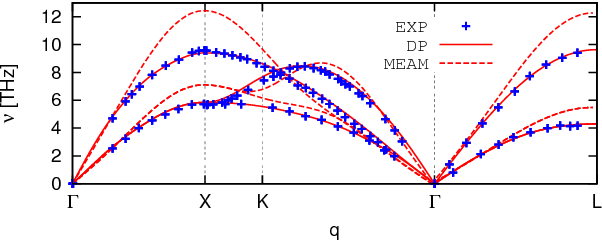
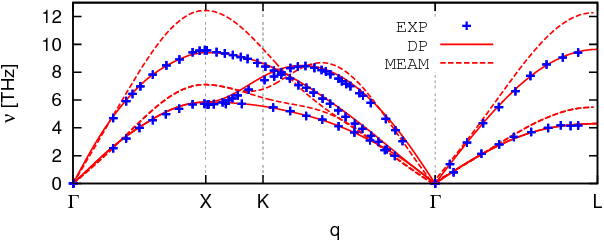
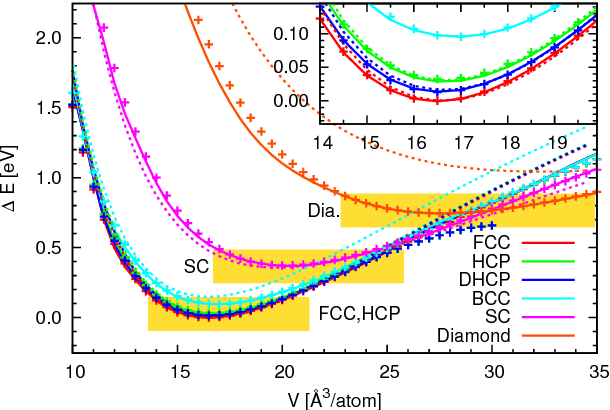
An active learning procedure called Deep Potential Generator (DP-GEN) is proposed for the construction of accurate and transferable machine learning-based models of the potential energy surface (PES) for the molecular modeling of materials. This procedure consists of three main components: exploration, labeling, and training. Application to the sample systems of Al, Mg and Al-Mg alloys demonstrates that DP-GEN can generate uniformly accurate PES models with a minimum number of labeled data.
Deep Potential Molecular Dynamics: a scalable model with the accuracy of quantum mechanics
Dec 12, 2017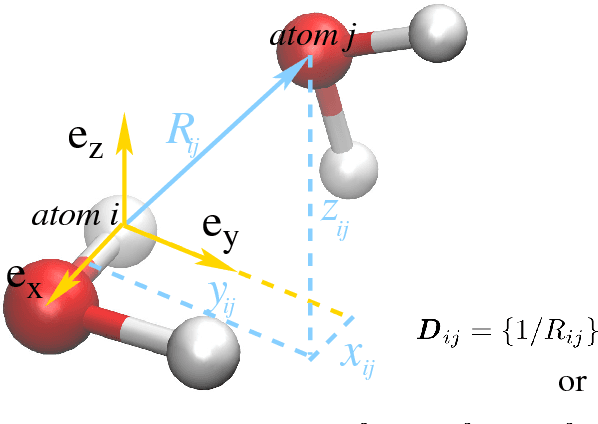

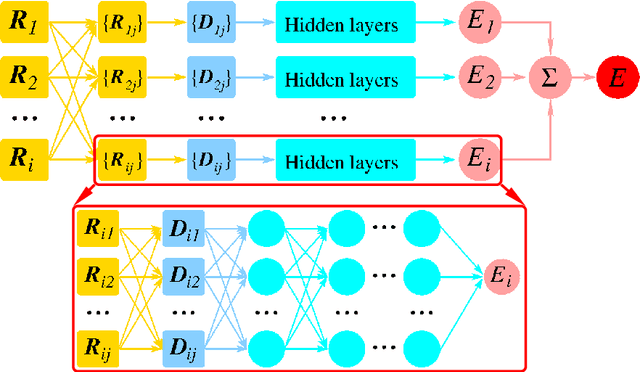
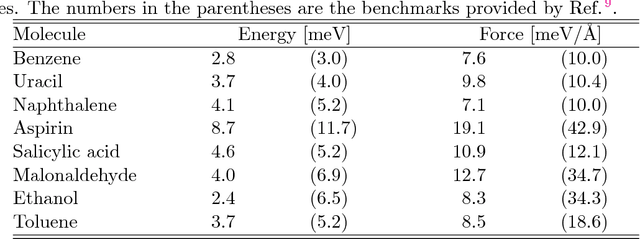
We introduce a scheme for molecular simulations, the Deep Potential Molecular Dynamics (DeePMD) method, based on a many-body potential and interatomic forces generated by a carefully crafted deep neural network trained with ab initio data. The neural network model preserves all the natural symmetries in the problem. It is "first principle-based" in the sense that there are no ad hoc components aside from the network model. We show that the proposed scheme provides an efficient and accurate protocol in a variety of systems, including bulk materials and molecules. In all these cases, DeePMD gives results that are essentially indistinguishable from the original data, at a cost that scales linearly with system size.