 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeepVRegulome: DNABERT-based deep-learning framework for predicting the functional impact of short genomic variants on the human regulome
Nov 12, 2025



Whole-genome sequencing (WGS) has revealed numerous non-coding short variants whose functional impacts remain poorly understood. Despite recent advances in deep-learning genomic approaches, accurately predicting and prioritizing clinically relevant mutations in gene regulatory regions remains a major challenge. Here we introduce Deep VRegulome, a deep-learning method for prediction and interpretation of functionally disruptive variants in the human regulome, which combines 700 DNABERT fine-tuned models, trained on vast amounts of ENCODE gene regulatory regions, with variant scoring, motif analysis, attention-based visualization, and survival analysis. We showcase its application on TCGA glioblastoma WGS dataset in prioritizing survival-associated mutations and regulatory regions. The analysis identified 572 splice-disrupting and 9,837 transcription-factor binding site altering mutations occurring in greater than 10% of glioblastoma samples. Survival analysis linked 1352 mutations and 563 disrupted regulatory regions to patient outcomes, enabling stratification via non-coding mutation signatures. All the code, fine-tuned models, and an interactive data portal are publicly available.
TFBS-Finder: Deep Learning-based Model with DNABERT and Convolutional Networks to Predict Transcription Factor Binding Sites
Feb 03, 2025



Transcription factors are proteins that regulate the expression of genes by binding to specific genomic regions known as Transcription Factor Binding Sites (TFBSs), typically located in the promoter regions of those genes. Accurate prediction of these binding sites is essential for understanding the complex gene regulatory networks underlying various cellular functions. In this regard, many deep learning models have been developed for such prediction, but there is still scope of improvement. In this work, we have developed a deep learning model which uses pre-trained DNABERT, a Convolutional Neural Network (CNN) module, a Modified Convolutional Block Attention Module (MCBAM), a Multi-Scale Convolutions with Attention (MSCA) module and an output module. The pre-trained DNABERT is used for sequence embedding, thereby capturing the long-term dependencies in the DNA sequences while the CNN, MCBAM and MSCA modules are useful in extracting higher-order local features. TFBS-Finder is trained and tested on 165 ENCODE ChIP-seq datasets. We have also performed ablation studies as well as cross-cell line validations and comparisons with other models. The experimental results show the superiority of the proposed method in predicting TFBSs compared to the existing methodologies. The codes and the relevant datasets are publicly available at https://github.com/NimishaGhosh/TFBS-Finder/.
DNABERT-2: Efficient Foundation Model and Benchmark For Multi-Species Genome
Jun 26, 2023



Decoding the linguistic intricacies of the genome is a crucial problem in biology, and pre-trained foundational models such as DNABERT and Nucleotide Transformer have made significant strides in this area. Existing works have largely hinged on k-mer, fixed-length permutations of A, T, C, and G, as the token of the genome language due to its simplicity. However, we argue that the computation and sample inefficiencies introduced by k-mer tokenization are primary obstacles in developing large genome foundational models. We provide conceptual and empirical insights into genome tokenization, building on which we propose to replace k-mer tokenization with Byte Pair Encoding (BPE), a statistics-based data compression algorithm that constructs tokens by iteratively merging the most frequent co-occurring genome segment in the corpus. We demonstrate that BPE not only overcomes the limitations of k-mer tokenization but also benefits from the computational efficiency of non-overlapping tokenization. Based on these insights, we introduce DNABERT-2, a refined genome foundation model that adapts an efficient tokenizer and employs multiple strategies to overcome input length constraints, reduce time and memory expenditure, and enhance model capability. Furthermore, we identify the absence of a comprehensive and standardized benchmark for genome understanding as another significant impediment to fair comparative analysis. In response, we propose the Genome Understanding Evaluation (GUE), a comprehensive multi-species genome classification dataset that amalgamates $28$ distinct datasets across $7$ tasks, with input lengths ranging from $70$ to $1000$. Through comprehensive experiments on the GUE benchmark, we demonstrate that DNABERT-2 achieves comparable performance to the state-of-the-art model with $21 \times$ fewer parameters and approximately $56 \times$ less GPU time in pre-training.
Multimodal Graph-based Transformer Framework for Biomedical Relation Extraction
Jul 01, 2021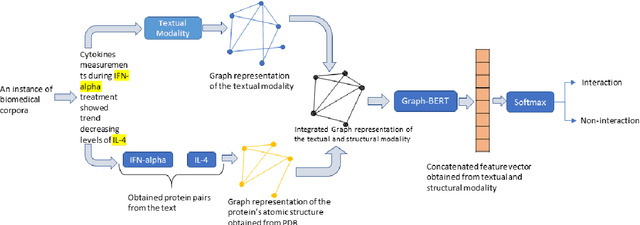
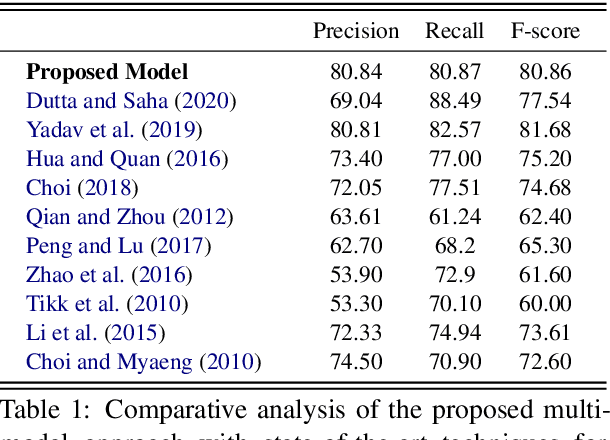
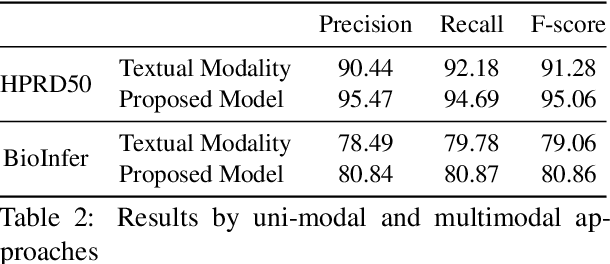
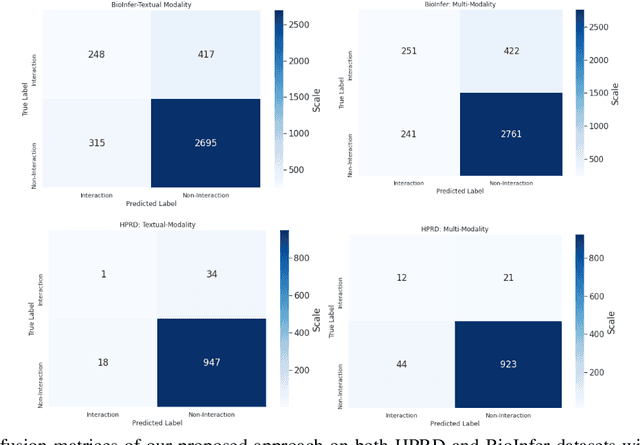
The recent advancement of pre-trained Transformer models has propelled the development of effective text mining models across various biomedical tasks. However, these models are primarily learned on the textual data and often lack the domain knowledge of the entities to capture the context beyond the sentence. In this study, we introduced a novel framework that enables the model to learn multi-omnics biological information about entities (proteins) with the help of additional multi-modal cues like molecular structure. Towards this, rather developing modality-specific architectures, we devise a generalized and optimized graph based multi-modal learning mechanism that utilizes the GraphBERT model to encode the textual and molecular structure information and exploit the underlying features of various modalities to enable end-to-end learning. We evaluated our proposed method on ProteinProtein Interaction task from the biomedical corpus, where our proposed generalized approach is observed to be benefited by the additional domain-specific modality.