 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDistribution-Conditioned Transport
Mar 05, 2026Learning a transport model that maps a source distribution to a target distribution is a canonical problem in machine learning, but scientific applications increasingly require models that can generalize to source and target distributions unseen during training. We introduce distribution-conditioned transport (DCT), a framework that conditions transport maps on learned embeddings of source and target distributions, enabling generalization to unseen distribution pairs. DCT also allows semi-supervised learning for distributional forecasting problems: because it learns from arbitrary distribution pairs, it can leverage distributions observed at only one condition to improve transport prediction. DCT is agnostic to the underlying transport mechanism, supporting models ranging from flow matching to distributional divergence-based models (e.g. Wasserstein, MMD). We demonstrate the practical performance benefits of DCT on synthetic benchmarks and four applications in biology: batch effect transfer in single-cell genomics, perturbation prediction from mass cytometry data, learning clonal transcriptional dynamics in hematopoiesis, and modeling T-cell receptor sequence evolution.
Count Bridges enable Modeling and Deconvolving Transcriptomic Data
Mar 05, 2026Many modern biological assays, including RNA sequencing, yield integer-valued counts that reflect the number of molecules detected. These measurements are often not at the desired resolution: while the unit of interest is typically a single cell, many measurement technologies produce counts aggregated over sets of cells. Although recent generative frameworks such as diffusion and flow matching have been extended to non-Euclidean and discrete settings, it remains unclear how best to model integer-valued data or how to systematically deconvolve aggregated observations. We introduce Count Bridges, a stochastic bridge process on the integers that provides an exact, tractable analogue of diffusion-style models for count data, with closed-form conditionals for efficient training and sampling. We extend this framework to enable direct training from aggregated measurements via an Expectation-Maximization-style approach that treats unit-level counts as latent variables. We demonstrate state-of-the-art performance on integer distribution matching benchmarks, comparing against flow matching and discrete flow matching baselines across various metrics. We then apply Count Bridges to two large-scale problems in biology: modeling single-cell gene expression data at the nucleotide resolution, with applications to deconvolving bulk RNA-seq, and resolving multicellular spatial transcriptomic spots into single-cell count profiles. Our methods offer a principled foundation for generative modeling and deconvolution of biological count data across scales and modalities.
Generative Distribution Embeddings
May 23, 2025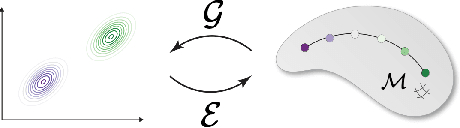

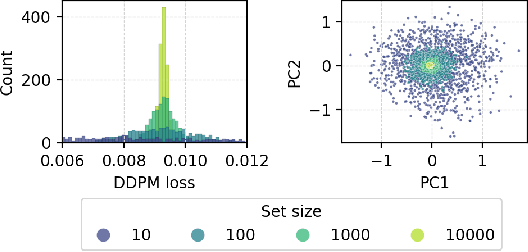
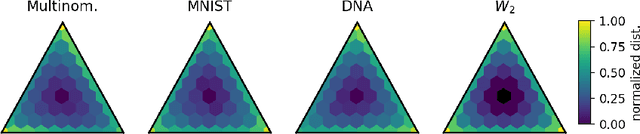
Many real-world problems require reasoning across multiple scales, demanding models which operate not on single data points, but on entire distributions. We introduce generative distribution embeddings (GDE), a framework that lifts autoencoders to the space of distributions. In GDEs, an encoder acts on sets of samples, and the decoder is replaced by a generator which aims to match the input distribution. This framework enables learning representations of distributions by coupling conditional generative models with encoder networks which satisfy a criterion we call distributional invariance. We show that GDEs learn predictive sufficient statistics embedded in the Wasserstein space, such that latent GDE distances approximately recover the $W_2$ distance, and latent interpolation approximately recovers optimal transport trajectories for Gaussian and Gaussian mixture distributions. We systematically benchmark GDEs against existing approaches on synthetic datasets, demonstrating consistently stronger performance. We then apply GDEs to six key problems in computational biology: learning representations of cell populations from lineage-tracing data (150K cells), predicting perturbation effects on single-cell transcriptomes (1M cells), predicting perturbation effects on cellular phenotypes (20M single-cell images), modeling tissue-specific DNA methylation patterns (253M sequences), designing synthetic yeast promoters (34M sequences), and spatiotemporal modeling of viral protein sequences (1M sequences).