 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultitask learning with semiempirical orbital charges enables sample-efficient MLIPs
May 22, 2026Machine learning interatomic potentials (MLIPs) require generating computationally expensive, large-scale training datasets to accurately simulate materials and molecules. Incorporating electronic structure information using multitask learning improves sample efficiency, however, training on full Hamiltonian matrices, which scale quadratically with the number of atoms, is intractable for large datasets. In this work, we show that multitask learning utilizing orbitally resolved semiempirical charges significantly improves sample efficiency and accuracy in MLIPs. To efficiently predict orbital charges, we implement a specialized equivariant model, reducing charge prediction error compared to an invariant baseline. By augmenting training with computationally inexpensive GFN1-xTB orbital charges, which scale linearly with the number of atoms, our model achieves a 46\% reduction in energy mean absolute error and requires five times less data to match the performance of energy-only models. Furthermore, our approach outperforms models trained on expensive density functional theory (DFT) atomic charges, capturing orbitally resolved electronic complexity and forcing the network to learn a physically accurate latent space that spontaneously clusters metals by shared chemical properties. Because orbital charges are only required during training, this approach preserves inference efficiency, providing a scalable recipe for developing accurate, data-efficient foundation models for complex chemical systems.
Using GNN property predictors as molecule generators
Jun 05, 2024



Graph neural networks (GNNs) have emerged as powerful tools to accurately predict materials and molecular properties in computational discovery pipelines. In this article, we exploit the invertible nature of these neural networks to directly generate molecular structures with desired electronic properties. Starting from a random graph or an existing molecule, we perform a gradient ascent while holding the GNN weights fixed in order to optimize its input, the molecular graph, towards the target property. Valence rules are enforced strictly through a judicious graph construction. The method relies entirely on the property predictor; no additional training is required on molecular structures. We demonstrate the application of this method by generating molecules with specific DFT-verified energy gaps and octanol-water partition coefficients (logP). Our approach hits target properties with rates comparable to or better than state-of-the-art generative models while consistently generating more diverse molecules.
Interpretable discovery of new semiconductors with machine learning
Jan 12, 2021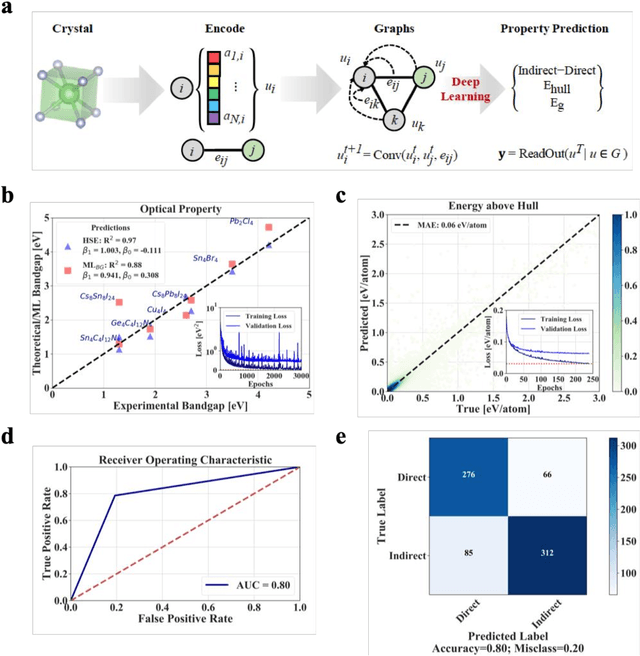
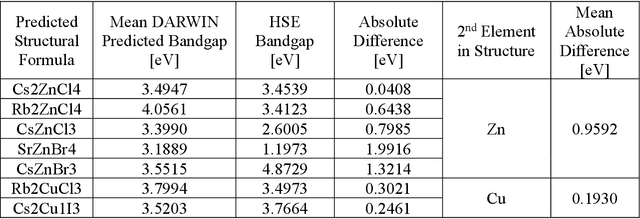
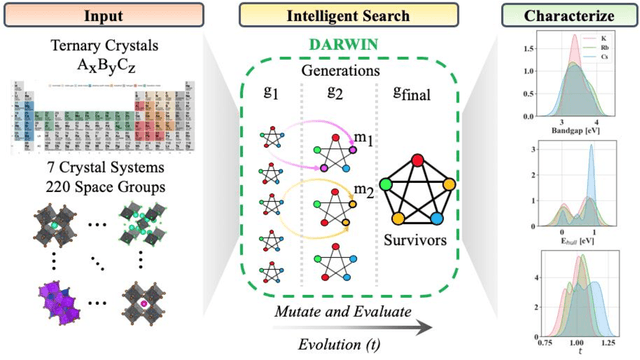
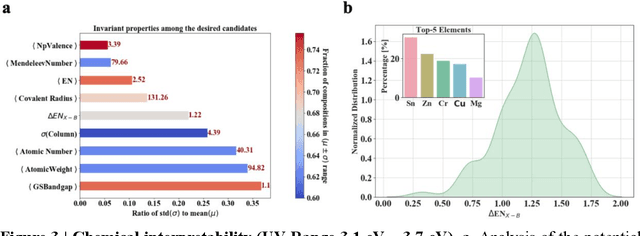
Machine learning models of materials$^{1-5}$ accelerate discovery compared to ab initio methods: deep learning models now reproduce density functional theory (DFT)-calculated results at one hundred thousandths of the cost of DFT$^{6}$. To provide guidance in experimental materials synthesis, these need to be coupled with an accurate yet effective search algorithm and training data consistent with experimental observations. Here we report an evolutionary algorithm powered search which uses machine-learned surrogate models trained on high-throughput hybrid functional DFT data benchmarked against experimental bandgaps: Deep Adaptive Regressive Weighted Intelligent Network (DARWIN). The strategy enables efficient search over the materials space of ~10$^8$ ternaries and 10$^{11}$ quaternaries$^{7}$ for candidates with target properties. It provides interpretable design rules, such as our finding that the difference in the electronegativity between the halide and B-site cation being a strong predictor of ternary structural stability. As an example, when we seek UV emission, DARWIN predicts K$_2$CuX$_3$ (X = Cl, Br) as a promising materials family, based on its electronegativity difference. We synthesized and found these materials to be stable, direct bandgap UV emitters. The approach also allows knowledge distillation for use by humans.