 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRare-Allele Detection Using Compressed Se(que)nsing
Sep 02, 2009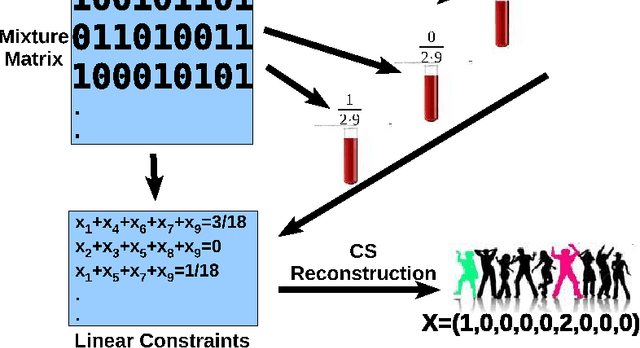
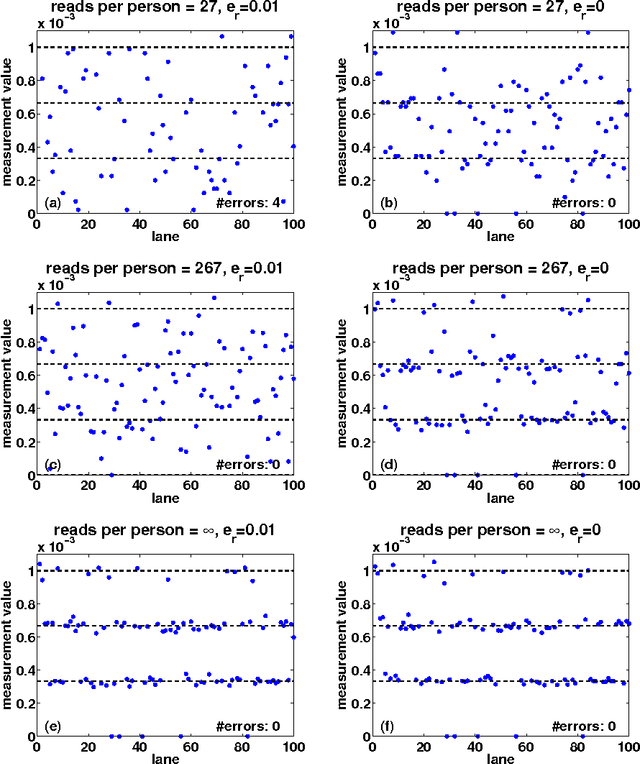
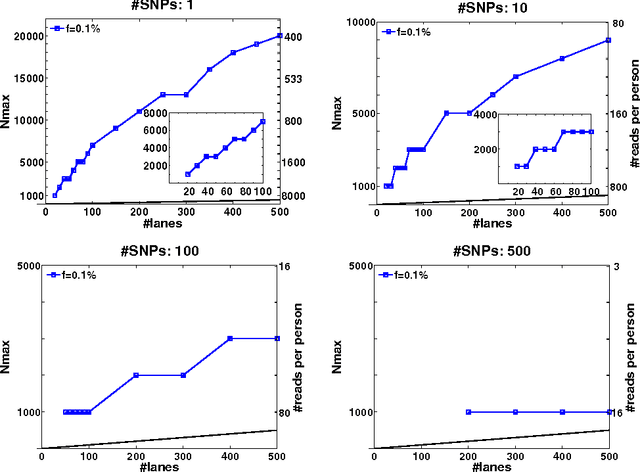
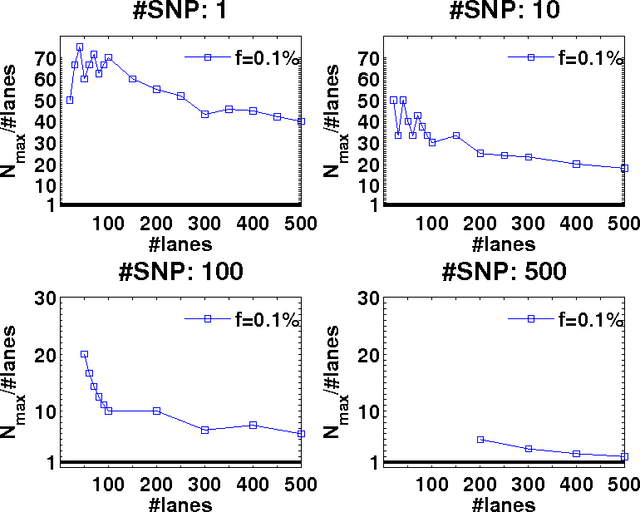
Detection of rare variants by resequencing is important for the identification of individuals carrying disease variants. Rapid sequencing by new technologies enables low-cost resequencing of target regions, although it is still prohibitive to test more than a few individuals. In order to improve cost trade-offs, it has recently been suggested to apply pooling designs which enable the detection of carriers of rare alleles in groups of individuals. However, this was shown to hold only for a relatively low number of individuals in a pool, and requires the design of pooling schemes for particular cases. We propose a novel pooling design, based on a compressed sensing approach, which is both general, simple and efficient. We model the experimental procedure and show via computer simulations that it enables the recovery of rare allele carriers out of larger groups than were possible before, especially in situations where high coverage is obtained for each individual. Our approach can also be combined with barcoding techniques to enhance performance and provide a feasible solution based on current resequencing costs. For example, when targeting a small enough genomic region (~100 base-pairs) and using only ~10 sequencing lanes and ~10 distinct barcodes, one can recover the identity of 4 rare allele carriers out of a population of over 4000 individuals.
Semi-Supervised Learning -- A Statistical Physics Approach
Apr 06, 2006



We present a novel approach to semi-supervised learning which is based on statistical physics. Most of the former work in the field of semi-supervised learning classifies the points by minimizing a certain energy function, which corresponds to a minimal k-way cut solution. In contrast to these methods, we estimate the distribution of classifications, instead of the sole minimal k-way cut, which yields more accurate and robust results. Our approach may be applied to all energy functions used for semi-supervised learning. The method is based on sampling using a Multicanonical Markov chain Monte-Carlo algorithm, and has a straightforward probabilistic interpretation, which allows for soft assignments of points to classes, and also to cope with yet unseen class types. The suggested approach is demonstrated on a toy data set and on two real-life data sets of gene expression.