 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhancing molecular dynamics with equivariant machine-learned densities
Apr 27, 2026Machine-learning interatomic potentials (MLIPs) have enabled molecular dynamics at near ab initio accuracy, yet remain limited to energies and forces by construction, leaving electronic observables such as dipole moments and polarizabilities inaccessible. We introduce DenSNet, a density-first approach to machine-learned electronic structure that learns the Hohenberg--Kohn map from nuclear configurations to the ground-state electron density. Our approach employs an SE(3)-equivariant neural network to predict density coefficients of a flexible atom-centered Gaussian basis, combined with a $Δ$-learning strategy that uses superposed atomic densities as a prior to accelerate training. A second equivariant network then maps the predicted density to the total energy, providing a unified framework for molecular dynamics and electronic structure. We validate DenSNet on ethanol, ethanethiol, and resorcinol, where infrared spectra from machine-learned trajectories show excellent agreement with experimental gas-phase measurements. To test scalability, we train on polythiophene oligomers with 1--6 monomers and extrapolate to chains of up to 12 monomers, generating stable long-time trajectories whose infrared spectra agree with reference density functional theory calculations. Here, we show that reinstating the electron density as the central learned quantity opens a practical route to transferable prediction of spectroscopic and electronic observables in large-scale molecular simulations.
Supervised Learning and the Finite-Temperature String Method for Computing Committor Functions and Reaction Rates
Jul 28, 2021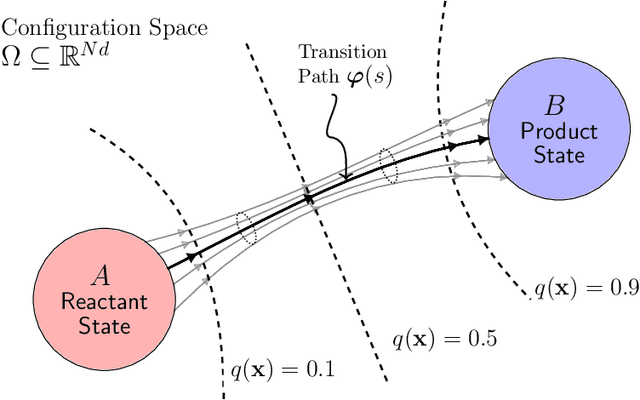


A central object in the computational studies of rare events is the committor function. Though costly to compute, the committor function encodes complete mechanistic information of the processes involving rare events, including reaction rates and transition-state ensembles. Under the framework of transition path theory (TPT), recent work [1] proposes an algorithm where a feedback loop couples a neural network that models the committor function with importance sampling, mainly umbrella sampling, which collects data needed for adaptive training. In this work, we show additional modifications are needed to improve the accuracy of the algorithm. The first modification adds elements of supervised learning, which allows the neural network to improve its prediction by fitting to sample-mean estimates of committor values obtained from short molecular dynamics trajectories. The second modification replaces the committor-based umbrella sampling with the finite-temperature string (FTS) method, which enables homogeneous sampling in regions where transition pathways are located. We test our modifications on low-dimensional systems with non-convex potential energy where reference solutions can be found via analytical or the finite element methods, and show how combining supervised learning and the FTS method yields accurate computation of committor functions and reaction rates. We also provide an error analysis for algorithms that use the FTS method, using which reaction rates can be accurately estimated during training with a small number of samples.