 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnraveling the Molecular Magic: AI Insights on the Formation of Extraordinarily Stretchable Hydrogels
Mar 08, 2024



The deliberate manipulation of ammonium persulfate, methylenebisacrylamide, dimethyleacrylamide, and polyethylene oxide concentrations resulted in the development of a hydrogel with an exceptional stretchability, capable of extending up to 260 times its original length. This study aims to elucidate the molecular architecture underlying this unique phenomenon by exploring potential reaction mechanisms, facilitated by an artificial intelligence prediction system. Artificial intelligence predictor introduces a novel approach to interlinking two polymers, involving the formation of networks interconnected with linear chains following random chain scission. This novel configuration leads to the emergence of a distinct type of hydrogel, herein referred to as a "Span Network." Additionally, Fourier-transform infrared spectroscopy (FTIR) is used to investigate functional groups that may be implicated in the proposed mechanism, with ester formation confirmed among numerous hydroxyl end groups obtained from chain scission of PEO and carboxyl groups formed on hydrogel networks.
AI for Interpretable Chemistry: Predicting Radical Mechanistic Pathways via Contrastive Learning
Nov 02, 2023



Deep learning-based reaction predictors have undergone significant architectural evolution. However, their reliance on reactions from the US Patent Office results in a lack of interpretable predictions and limited generalization capability to other chemistry domains, such as radical and atmospheric chemistry. To address these challenges, we introduce a new reaction predictor system, RMechRP, that leverages contrastive learning in conjunction with mechanistic pathways, the most interpretable representation of chemical reactions. Specifically designed for radical reactions, RMechRP provides different levels of interpretation of chemical reactions. We develop and train multiple deep-learning models using RMechDB, a public database of radical reactions, to establish the first benchmark for predicting radical reactions. Our results demonstrate the effectiveness of RMechRP in providing accurate and interpretable predictions of radical reactions, and its potential for various applications in atmospheric chemistry.
Deep Learning Models of the Discrete Component of the Galactic Interstellar Gamma-Ray Emission
Jun 06, 2022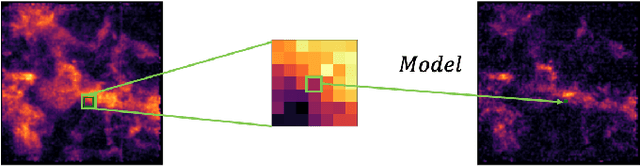
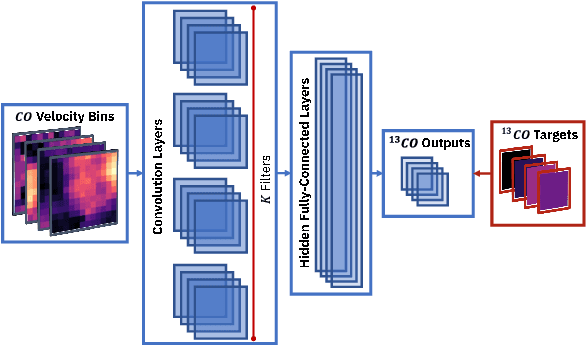

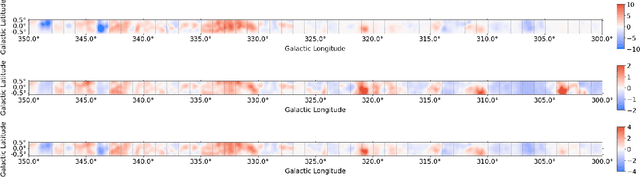
A significant point-like component from the small scale (or discrete) structure in the H2 interstellar gas might be present in the Fermi-LAT data, but modeling this emission relies on observations of rare gas tracers only available in limited regions of the sky. Identifying this contribution is important to discriminate gamma-ray point sources from interstellar gas, and to better characterize extended gamma-ray sources. We design and train convolutional neural networks to predict this emission where observations of these rare tracers do not exist and discuss the impact of this component on the analysis of the Fermi-LAT data. In particular, we evaluate prospects to exploit this methodology in the characterization of the Fermi-LAT Galactic center excess through accurate modeling of point-like structures in the data to help distinguish between a point-like or smooth nature for the excess. We show that deep learning may be effectively employed to model the gamma-ray emission traced by these rare H2 proxies within statistical significance in data-rich regions, supporting prospects to employ these methods in yet unobserved regions.
Rxn Hypergraph: a Hypergraph Attention Model for Chemical Reaction Representation
Jan 02, 2022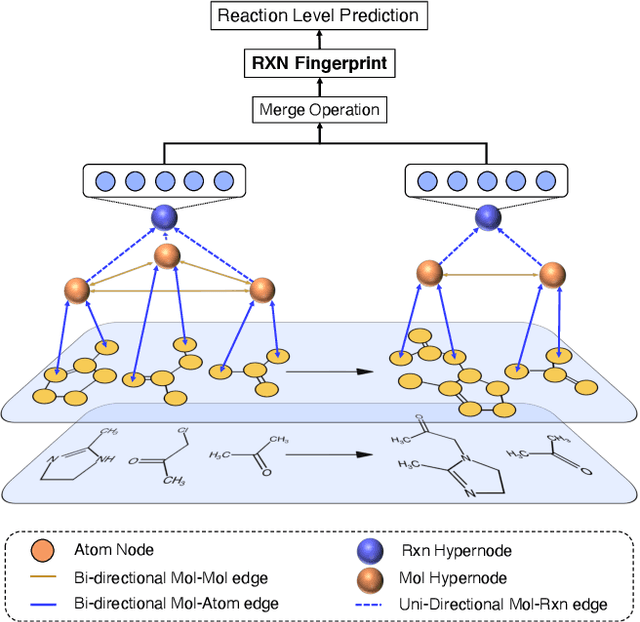
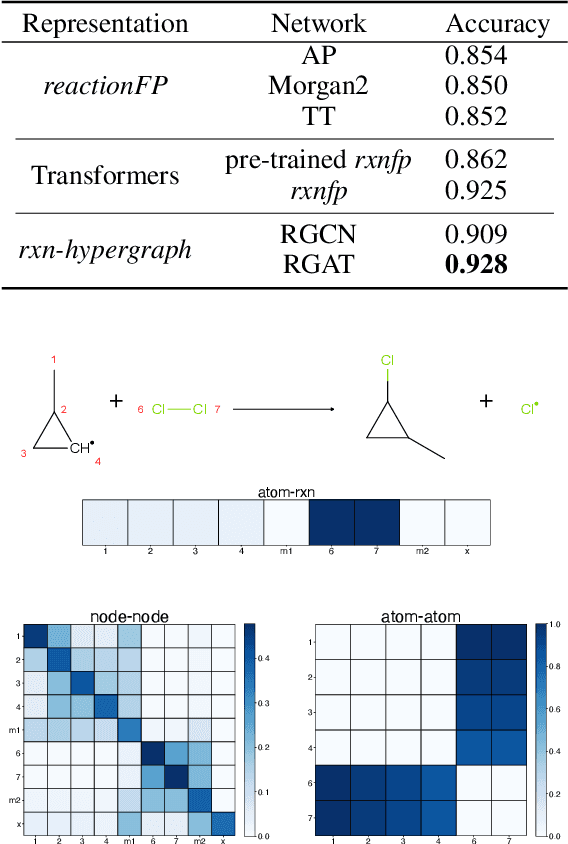
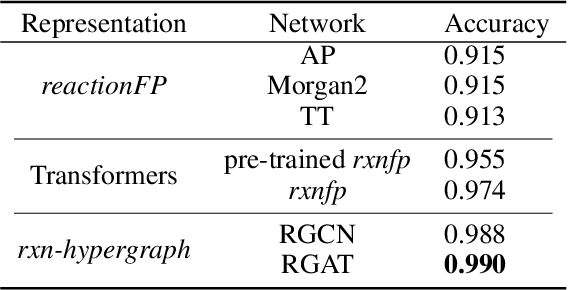
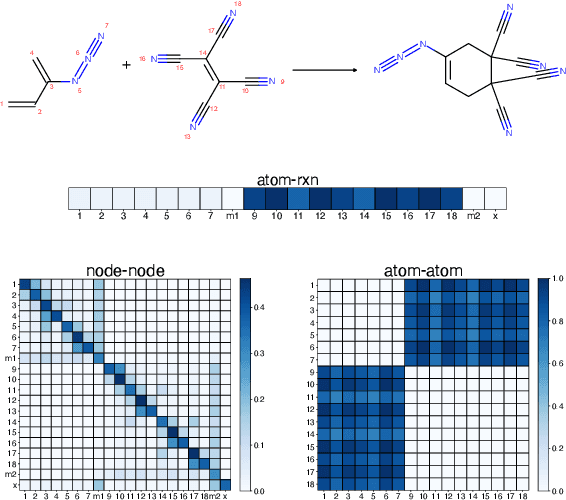
It is fundamental for science and technology to be able to predict chemical reactions and their properties. To achieve such skills, it is important to develop good representations of chemical reactions, or good deep learning architectures that can learn such representations automatically from the data. There is currently no universal and widely adopted method for robustly representing chemical reactions. Most existing methods suffer from one or more drawbacks, such as: (1) lacking universality; (2) lacking robustness; (3) lacking interpretability; or (4) requiring excessive manual pre-processing. Here we exploit graph-based representations of molecular structures to develop and test a hypergraph attention neural network approach to solve at once the reaction representation and property-prediction problems, alleviating the aforementioned drawbacks. We evaluate this hypergraph representation in three experiments using three independent data sets of chemical reactions. In all experiments, the hypergraph-based approach matches or outperforms other representations and their corresponding models of chemical reactions while yielding interpretable multi-level representations.
Tourbillon: a Physically Plausible Neural Architecture
Jul 22, 2021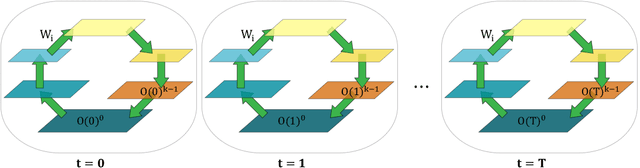
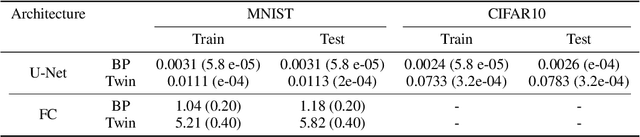
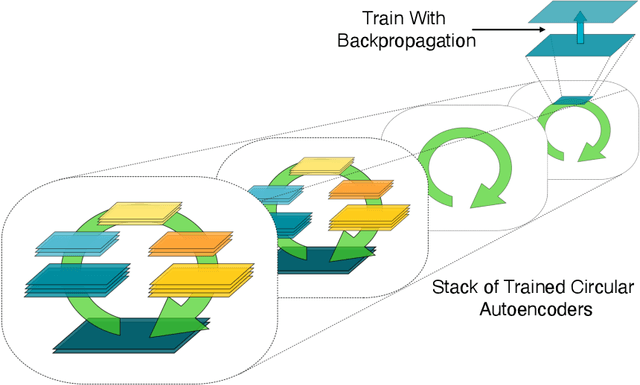
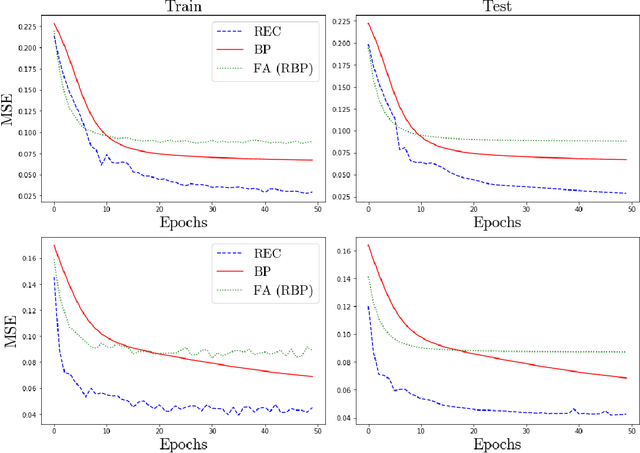
In a physical neural system, backpropagation is faced with a number of obstacles including: the need for labeled data, the violation of the locality learning principle, the need for symmetric connections, and the lack of modularity. Tourbillon is a new architecture that addresses all these limitations. At its core, it consists of a stack of circular autoencoders followed by an output layer. The circular autoencoders are trained in self-supervised mode by recirculation algorithms and the top layer in supervised mode by stochastic gradient descent, with the option of propagating error information through the entire stack using non-symmetric connections. While the Tourbillon architecture is meant primarily to address physical constraints, and not to improve current engineering applications of deep learning, we demonstrate its viability on standard benchmark datasets including MNIST, Fashion MNIST, and CIFAR10. We show that Tourbillon can achieve comparable performance to models trained with backpropagation and outperform models that are trained with other physically plausible algorithms, such as feedback alignment.
Quantum Mechanics and Machine Learning Synergies: Graph Attention Neural Networks to Predict Chemical Reactivity
Mar 24, 2021
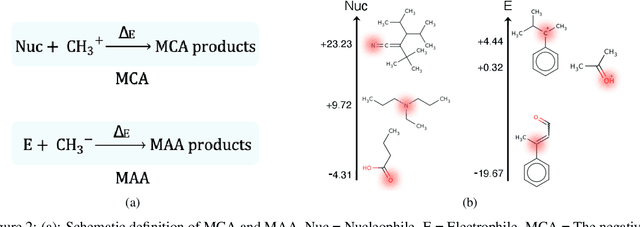
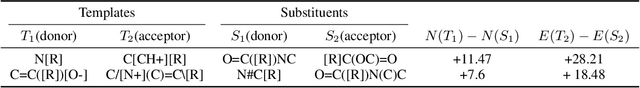
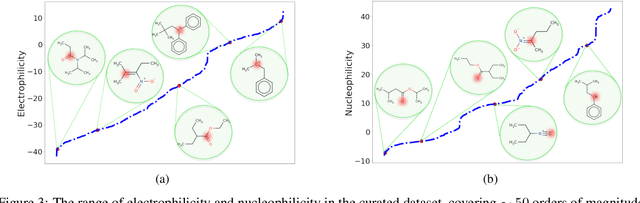
There is a lack of scalable quantitative measures of reactivity for functional groups in organic chemistry. Measuring reactivity experimentally is costly and time-consuming and does not scale to the astronomical size of chemical space. In previous quantum chemistry studies, we have introduced Methyl Cation Affinities (MCA*) and Methyl Anion Affinities (MAA*), using a solvation model, as quantitative measures of reactivity for organic functional groups over the broadest range. Although MCA* and MAA* offer good estimates of reactivity parameters, their calculation through Density Functional Theory (DFT) simulations is time-consuming. To circumvent this problem, we first use DFT to calculate MCA* and MAA* for more than 2,400 organic molecules thereby establishing a large dataset of chemical reactivity scores. We then design deep learning methods to predict the reactivity of molecular structures and train them using this curated dataset in combination with different representations of molecular structures. Using ten-fold cross-validation, we show that graph attention neural networks applied to informative input fingerprints produce the most accurate estimates of reactivity, achieving over 91% test accuracy for predicting the MCA* plus-minus 3.0 or MAA* plus-minus 3.0, over 50 orders of magnitude. Finally, we demonstrate the application of these reactivity scores to two tasks: (1) chemical reaction prediction; (2) combinatorial generation of reaction mechanisms. The curated dataset of MCA* and MAA* scores is available through the ChemDB chemoinformatics web portal at www.cdb.ics.uci.edu.
SPLASH: Learnable Activation Functions for Improving Accuracy and Adversarial Robustness
Jun 16, 2020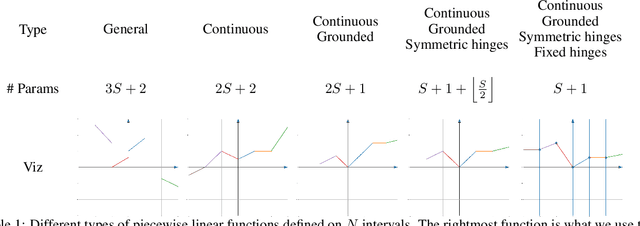
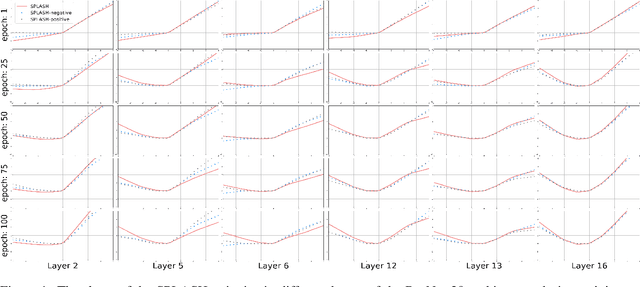
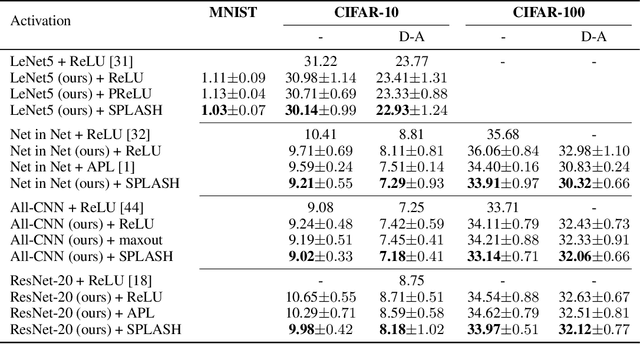
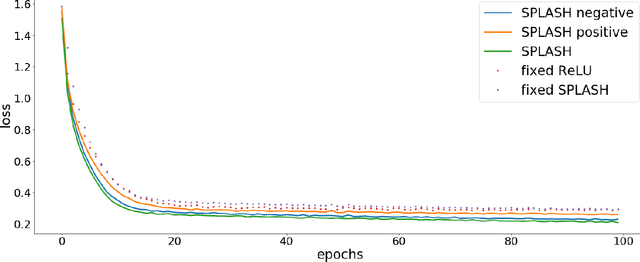
We introduce SPLASH units, a class of learnable activation functions shown to simultaneously improve the accuracy of deep neural networks while also improving their robustness to adversarial attacks. SPLASH units have both a simple parameterization and maintain the ability to approximate a wide range of non-linear functions. SPLASH units are: 1) continuous; 2) grounded (f(0) = 0); 3) use symmetric hinges; and 4) the locations of the hinges are derived directly from the data (i.e. no learning required). Compared to nine other learned and fixed activation functions, including ReLU and its variants, SPLASH units show superior performance across three datasets (MNIST, CIFAR-10, and CIFAR-100) and four architectures (LeNet5, All-CNN, ResNet-20, and Network-in-Network). Furthermore, we show that SPLASH units significantly increase the robustness of deep neural networks to adversarial attacks. Our experiments on both black-box and open-box adversarial attacks show that commonly-used architectures, namely LeNet5, All-CNN, ResNet-20, and Network-in-Network, can be up to 31% more robust to adversarial attacks by simply using SPLASH units instead of ReLUs.
Continuous Representation of Molecules Using Graph Variational Autoencoder
Apr 17, 2020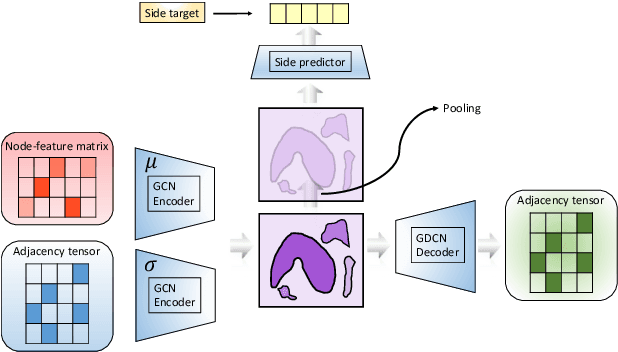
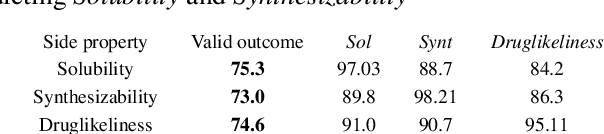
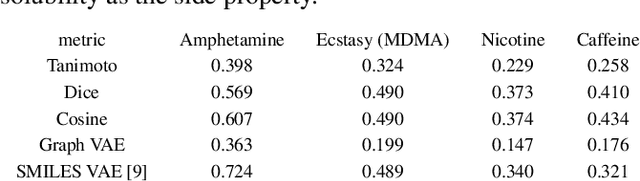
In order to continuously represent molecules, we propose a generative model in the form of a VAE which is operating on the 2D-graph structure of molecules. A side predictor is employed to prune the latent space and help the decoder in generating meaningful adjacency tensor of molecules. Other than the potential applicability in drug design and property prediction, we show the superior performance of this technique in comparison to other similar methods based on the SMILES representation of the molecules with RNN based encoder and decoder.