 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSolvaformer: an SE(3)-equivariant graph transformer for small molecule solubility prediction
Nov 12, 2025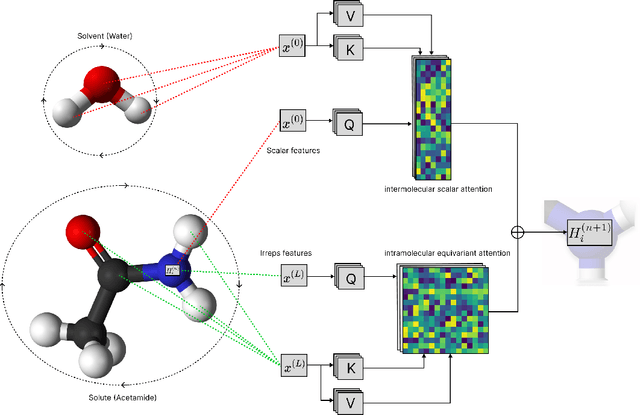
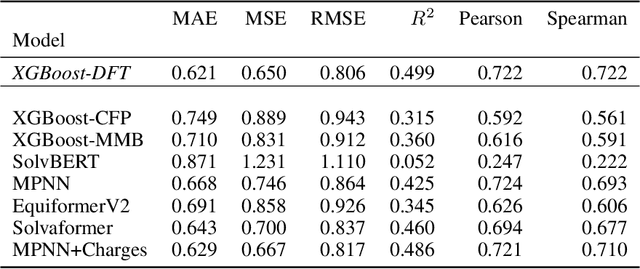
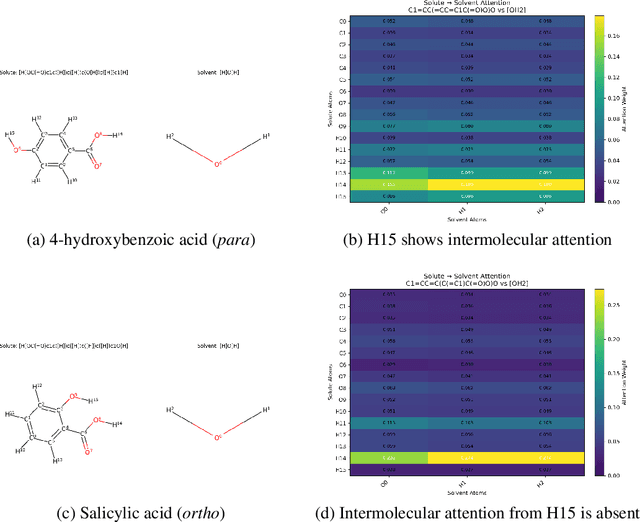

Accurate prediction of small molecule solubility using material-sparing approaches is critical for accelerating synthesis and process optimization, yet experimental measurement is costly and many learning approaches either depend on quantumderived descriptors or offer limited interpretability. We introduce Solvaformer, a geometry-aware graph transformer that models solutions as multiple molecules with independent SE(3) symmetries. The architecture combines intramolecular SE(3)-equivariant attention with intermolecular scalar attention, enabling cross-molecular communication without imposing spurious relative geometry. We train Solvaformer in a multi-task setting to predict both solubility (log S) and solvation free energy, using an alternating-batch regimen that trains on quantum-mechanical data (CombiSolv-QM) and on experimental measurements (BigSolDB 2.0). Solvaformer attains the strongest overall performance among the learned models and approaches a DFT-assisted gradient-boosting baseline, while outperforming an EquiformerV2 ablation and sequence-based alternatives. In addition, token-level attention produces chemically coherent attributions: case studies recover known intra- vs. inter-molecular hydrogen-bonding patterns that govern solubility differences in positional isomers. Taken together, Solvaformer provides an accurate, scalable, and interpretable approach to solution-phase property prediction by uniting geometric inductive bias with a mixed dataset training strategy on complementary computational and experimental data.
Many-Shot In-Context Learning for Molecular Inverse Design
Jul 26, 2024Large Language Models (LLMs) have demonstrated great performance in few-shot In-Context Learning (ICL) for a variety of generative and discriminative chemical design tasks. The newly expanded context windows of LLMs can further improve ICL capabilities for molecular inverse design and lead optimization. To take full advantage of these capabilities we developed a new semi-supervised learning method that overcomes the lack of experimental data available for many-shot ICL. Our approach involves iterative inclusion of LLM generated molecules with high predicted performance, along with experimental data. We further integrated our method in a multi-modal LLM which allows for the interactive modification of generated molecular structures using text instructions. As we show, the new method greatly improves upon existing ICL methods for molecular design while being accessible and easy to use for scientists.