 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMACS: Multi-Agent Reinforcement Learning for Optimization of Crystal Structures
Jun 04, 2025



Geometry optimization of atomic structures is a common and crucial task in computational chemistry and materials design. Following the learning to optimize paradigm, we propose a new multi-agent reinforcement learning method called Multi-Agent Crystal Structure optimization (MACS) to address periodic crystal structure optimization. MACS treats geometry optimization as a partially observable Markov game in which atoms are agents that adjust their positions to collectively discover a stable configuration. We train MACS across various compositions of reported crystalline materials to obtain a policy that successfully optimizes structures from the training compositions as well as structures of larger sizes and unseen compositions, confirming its excellent scalability and zero-shot transferability. We benchmark our approach against a broad range of state-of-the-art optimization methods and demonstrate that MACS optimizes periodic crystal structures significantly faster, with fewer energy calculations, and the lowest failure rate.
Assessing data-driven predictions of band gap and electrical conductivity for transparent conducting materials
Nov 21, 2024Machine Learning (ML) has offered innovative perspectives for accelerating the discovery of new functional materials, leveraging the increasing availability of material databases. Despite the promising advances, data-driven methods face constraints imposed by the quantity and quality of available data. Moreover, ML is often employed in tandem with simulated datasets originating from density functional theory (DFT), and assessed through in-sample evaluation schemes. This scenario raises questions about the practical utility of ML in uncovering new and significant material classes for industrial applications. Here, we propose a data-driven framework aimed at accelerating the discovery of new transparent conducting materials (TCMs), an important category of semiconductors with a wide range of applications. To mitigate the shortage of available data, we create and validate unique experimental databases, comprising several examples of existing TCMs. We assess state-of-the-art (SOTA) ML models for property prediction from the stoichiometry alone. We propose a bespoke evaluation scheme to provide empirical evidence on the ability of ML to uncover new, previously unseen materials of interest. We test our approach on a list of 55 compositions containing typical elements of known TCMs. Although our study indicates that ML tends to identify new TCMs compositionally similar to those in the training data, we empirically demonstrate that it can highlight material candidates that may have been previously overlooked, offering a systematic approach to identify materials that are likely to display TCMs characteristics.
Element selection for functional materials discovery by integrated machine learning of atomic contributions to properties
Feb 02, 2022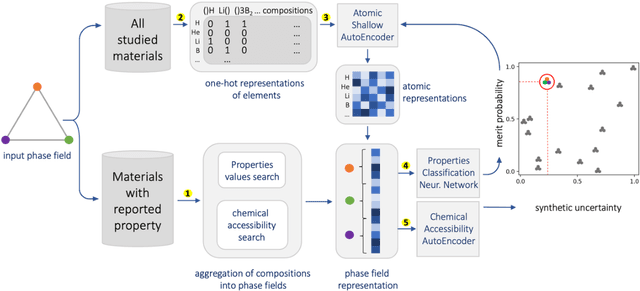
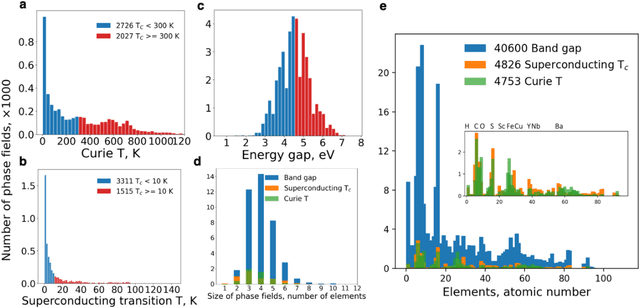
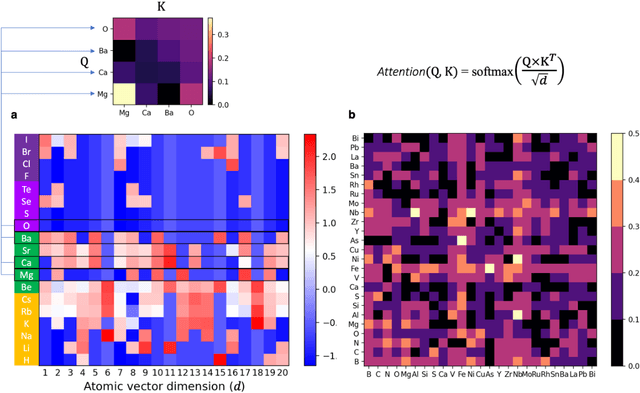
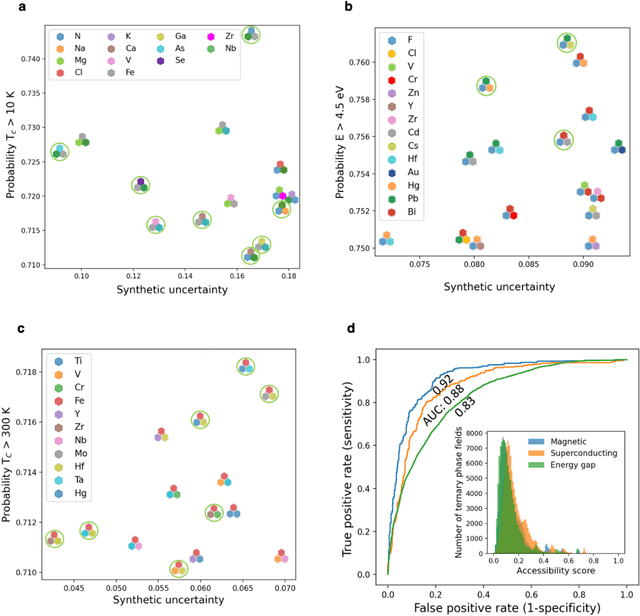
At the high level, the fundamental differences between materials originate from the unique nature of the constituent chemical elements. Before specific differences emerge according to the precise ratios of elements (composition) in a given crystal structure (phase), the material can be represented by its phase field defined simply as the set of the constituent chemical elements. Classification of the materials at the level of their phase fields can accelerate materials discovery by selecting the elemental combinations that are likely to produce desirable functional properties in synthetically accessible materials. Here, we demonstrate that classification of the materials phase field with respect to the maximum expected value of a target functional property can be combined with the ranking of the materials synthetic accessibility. This end-to-end machine learning approach (PhaseSelect) first derives the atomic characteristics from the compositional environments in all computationally and experimentally explored materials and then employs these characteristics to classify the phase field by their merit. PhaseSelect can quantify the materials potential at the level of the periodic table, which we demonstrate with significant accuracy for three avenues of materials applications: high-temperature superconducting, high-temperature magnetic and targetted energy band gap materials.