 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBoltzina: Efficient and Accurate Virtual Screening via Docking-Guided Binding Prediction with Boltz-2
Aug 24, 2025In structure-based drug discovery, virtual screening using conventional molecular docking methods can be performed rapidly but suffers from limitations in prediction accuracy. Recently, Boltz-2 was proposed, achieving extremely high accuracy in binding affinity prediction, but requiring approximately 20 seconds per compound per GPU, making it difficult to apply to large-scale screening of hundreds of thousands to millions of compounds. This study proposes Boltzina, a novel framework that leverages Boltz-2's high accuracy while significantly improving computational efficiency. Boltzina achieves both accuracy and speed by omitting the rate-limiting structure prediction from Boltz-2's architecture and directly predicting affinity from AutoDock Vina docking poses. We evaluate on eight assays from the MF-PCBA dataset and show that while Boltzina performs below Boltz-2, it provides significantly higher screening performance compared to AutoDock Vina and GNINA. Additionally, Boltzina achieved up to 11.8$\times$ faster through reduced recycling iterations and batch processing. Furthermore, we investigated multi-pose selection strategies and two-stage screening combining Boltzina and Boltz-2, presenting optimization methods for accuracy and efficiency according to application requirements. This study represents the first attempt to apply Boltz-2's high-accuracy predictions to practical-scale screening, offering a pipeline that combines both accuracy and efficiency in computational biology. The Boltzina is available on github; https://github.com/ohuelab/boltzina.
NPGPT: Natural Product-Like Compound Generation with GPT-based Chemical Language Models
Nov 19, 2024Natural products are substances produced by organisms in nature and often possess biological activity and structural diversity. Drug development based on natural products has been common for many years. However, the intricate structures of these compounds present challenges in terms of structure determination and synthesis, particularly compared to the efficiency of high-throughput screening of synthetic compounds. In recent years, deep learning-based methods have been applied to the generation of molecules. In this study, we trained chemical language models on a natural product dataset and generated natural product-like compounds. The results showed that the distribution of the compounds generated was similar to that of natural products. We also evaluated the effectiveness of the generated compounds as drug candidates. Our method can be used to explore the vast chemical space and reduce the time and cost of drug discovery of natural products.
Active learning for energy-based antibody optimization and enhanced screening
Sep 18, 2024Accurate prediction and optimization of protein-protein binding affinity is crucial for therapeutic antibody development. Although machine learning-based prediction methods $\Delta\Delta G$ are suitable for large-scale mutant screening, they struggle to predict the effects of multiple mutations for targets without existing binders. Energy function-based methods, though more accurate, are time consuming and not ideal for large-scale screening. To address this, we propose an active learning workflow that efficiently trains a deep learning model to learn energy functions for specific targets, combining the advantages of both approaches. Our method integrates the RDE-Network deep learning model with Rosetta's energy function-based Flex ddG to efficiently explore mutants. In a case study targeting HER2-binding Trastuzumab mutants, our approach significantly improved the screening performance over random selection and demonstrated the ability to identify mutants with better binding properties without experimental $\Delta\Delta G$ data. This workflow advances computational antibody design by combining machine learning, physics-based computations, and active learning to achieve more efficient antibody development.
Enhancing Model Learning and Interpretation Using Multiple Molecular Graph Representations for Compound Property and Activity Prediction
Apr 13, 2023Graph neural networks (GNNs) demonstrate great performance in compound property and activity prediction due to their capability to efficiently learn complex molecular graph structures. However, two main limitations persist including compound representation and model interpretability. While atom-level molecular graph representations are commonly used because of their ability to capture natural topology, they may not fully express important substructures or functional groups which significantly influence molecular properties. Consequently, recent research proposes alternative representations employing reduction techniques to integrate higher-level information and leverages both representations for model learning. However, there is still a lack of study about different molecular graph representations on model learning and interpretation. Interpretability is also crucial for drug discovery as it can offer chemical insights and inspiration for optimization. Numerous studies attempt to include model interpretation to explain the rationale behind predictions, but most of them focus solely on individual prediction with little analysis of the interpretation on different molecular graph representations. This research introduces multiple molecular graph representations that incorporate higher-level information and investigates their effects on model learning and interpretation from diverse perspectives. The results indicate that combining atom graph representation with reduced molecular graph representation can yield promising model performance. Furthermore, the interpretation results can provide significant features and potential substructures consistently aligning with background knowledge. These multiple molecular graph representations and interpretation analysis can bolster model comprehension and facilitate relevant applications in drug discovery.
Compound virtual screening by learning-to-rank with gradient boosting decision tree and enrichment-based cumulative gain
May 04, 2022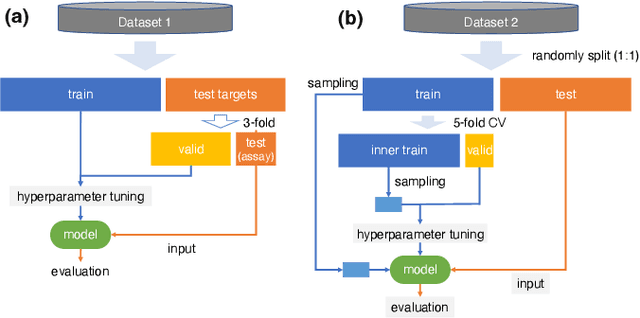
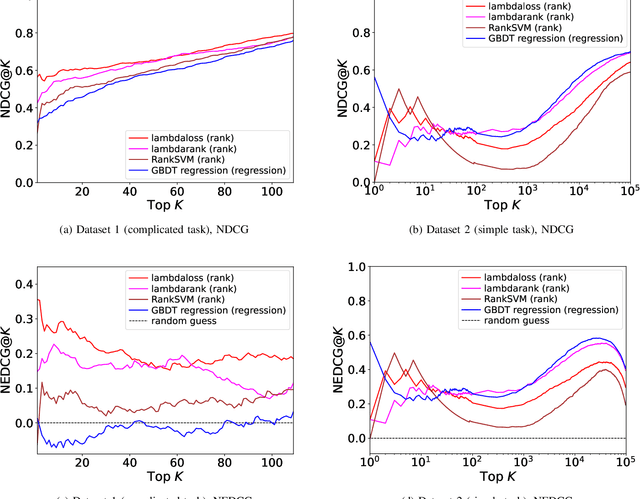
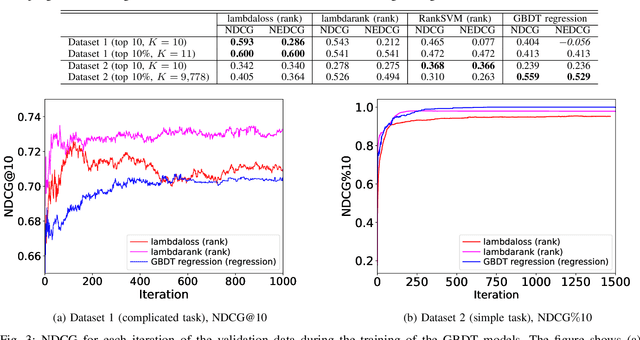
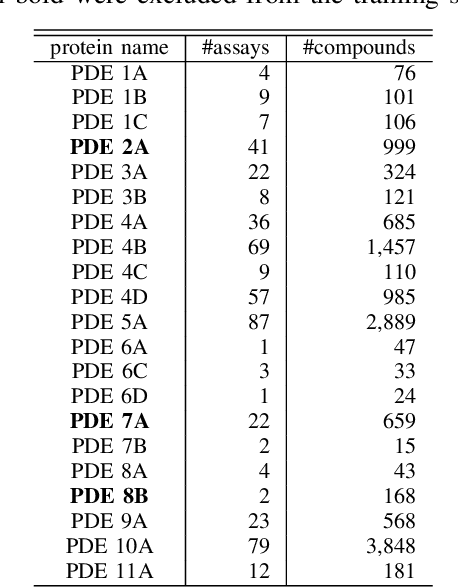
Learning-to-rank, a machine learning technique widely used in information retrieval, has recently been applied to the problem of ligand-based virtual screening, to accelerate the early stages of new drug development. Ranking prediction models learn based on ordinal relationships, making them suitable for integrating assay data from various environments. Existing studies of rank prediction in compound screening have generally used a learning-to-rank method called RankSVM. However, they have not been compared with or validated against the gradient boosting decision tree (GBDT)-based learning-to-rank methods that have gained popularity recently. Furthermore, although the ranking metric called Normalized Discounted Cumulative Gain (NDCG) is widely used in information retrieval, it only determines whether the predictions are better than those of other models. In other words, NDCG is incapable of recognizing when a prediction model produces worse than random results. Nevertheless, NDCG is still used in the performance evaluation of compound screening using learning-to-rank. This study used the GBDT model with ranking loss functions, called lambdarank and lambdaloss, for ligand-based virtual screening; results were compared with existing RankSVM methods and GBDT models using regression. We also proposed a new ranking metric, Normalized Enrichment Discounted Cumulative Gain (NEDCG), which aims to properly evaluate the goodness of ranking predictions. Results showed that the GBDT model with learning-to-rank outperformed existing regression methods using GBDT and RankSVM on diverse datasets. Moreover, NEDCG showed that predictions by regression were comparable to random predictions in multi-assay, multi-family datasets, demonstrating its usefulness for a more direct assessment of compound screening performance.
Molecular activity prediction using graph convolutional deep neural network considering distance on a molecular graph
Jul 04, 2019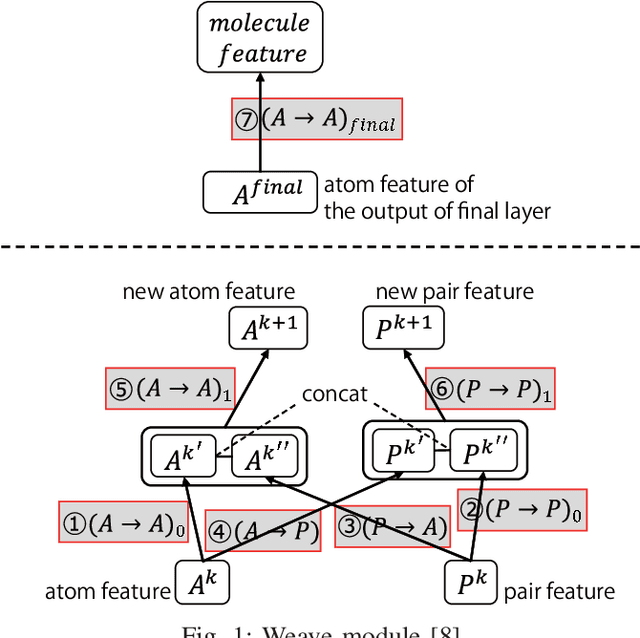
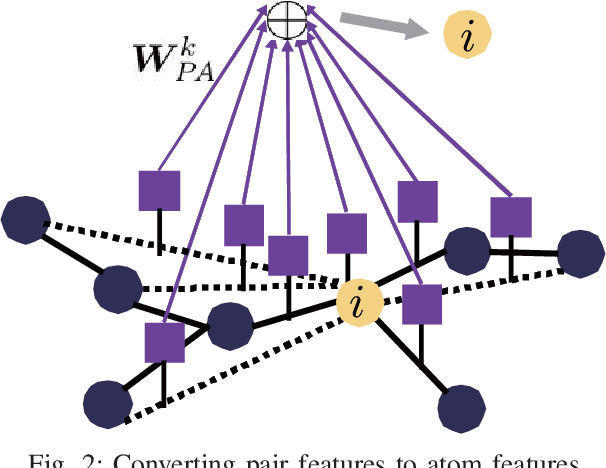

Machine learning is often used in virtual screening to find compounds that are pharmacologically active on a target protein. The weave module is a type of graph convolutional deep neural network that uses not only features focusing on atoms alone (atom features) but also features focusing on atom pairs (pair features); thus, it can consider information of nonadjacent atoms. However, the correlation between the distance on the graph and the three-dimensional coordinate distance is uncertain. In this paper, we propose three improvements for modifying the weave module. First, the distances between ring atoms on the graph were modified to bring the distances on the graph closer to the coordinate distance. Second, different weight matrices were used depending on the distance on the graph in the convolution layers of the pair features. Finally, a weighted sum, by distance, was used when converting pair features to atom features. The experimental results show that the performance of the proposed method is slightly better than that of the weave module, and the improvement in the distance representation might be useful for compound activity prediction.
Link Mining for Kernel-based Compound-Protein Interaction Predictions Using a Chemogenomics Approach
Jun 30, 2017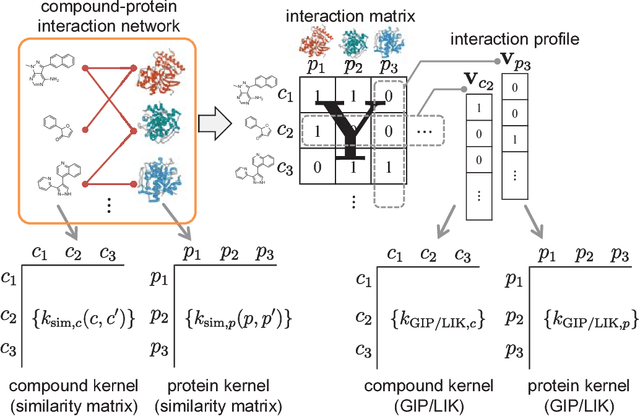
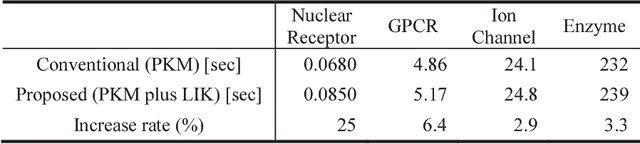
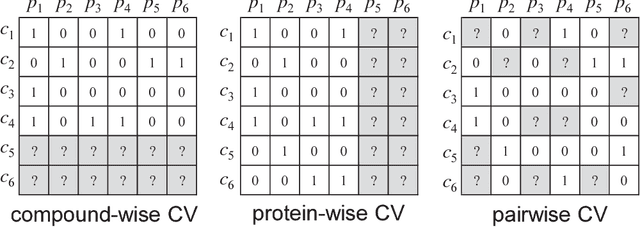
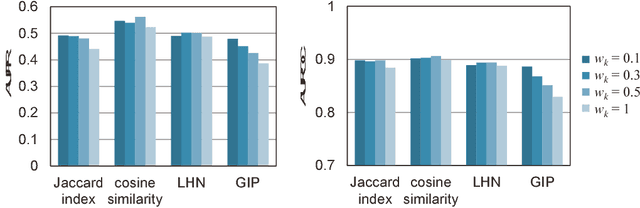
Virtual screening (VS) is widely used during computational drug discovery to reduce costs. Chemogenomics-based virtual screening (CGBVS) can be used to predict new compound-protein interactions (CPIs) from known CPI network data using several methods, including machine learning and data mining. Although CGBVS facilitates highly efficient and accurate CPI prediction, it has poor performance for prediction of new compounds for which CPIs are unknown. The pairwise kernel method (PKM) is a state-of-the-art CGBVS method and shows high accuracy for prediction of new compounds. In this study, on the basis of link mining, we improved the PKM by combining link indicator kernel (LIK) and chemical similarity and evaluated the accuracy of these methods. The proposed method obtained an average area under the precision-recall curve (AUPR) value of 0.562, which was higher than that achieved by the conventional Gaussian interaction profile (GIP) method (0.425), and the calculation time was only increased by a few percent.