 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular activity prediction using graph convolutional deep neural network considering distance on a molecular graph
Jul 04, 2019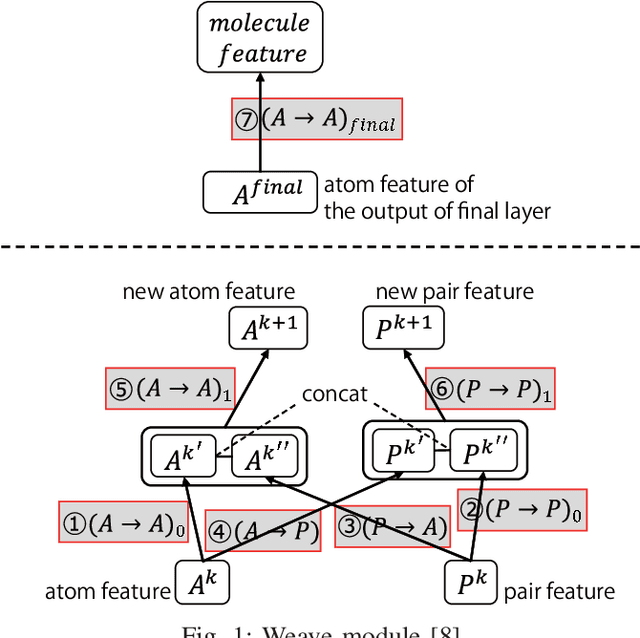
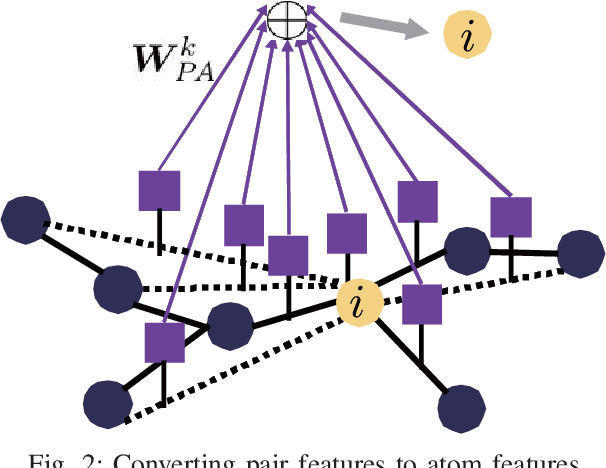

Machine learning is often used in virtual screening to find compounds that are pharmacologically active on a target protein. The weave module is a type of graph convolutional deep neural network that uses not only features focusing on atoms alone (atom features) but also features focusing on atom pairs (pair features); thus, it can consider information of nonadjacent atoms. However, the correlation between the distance on the graph and the three-dimensional coordinate distance is uncertain. In this paper, we propose three improvements for modifying the weave module. First, the distances between ring atoms on the graph were modified to bring the distances on the graph closer to the coordinate distance. Second, different weight matrices were used depending on the distance on the graph in the convolution layers of the pair features. Finally, a weighted sum, by distance, was used when converting pair features to atom features. The experimental results show that the performance of the proposed method is slightly better than that of the weave module, and the improvement in the distance representation might be useful for compound activity prediction.
Link Mining for Kernel-based Compound-Protein Interaction Predictions Using a Chemogenomics Approach
Jun 30, 2017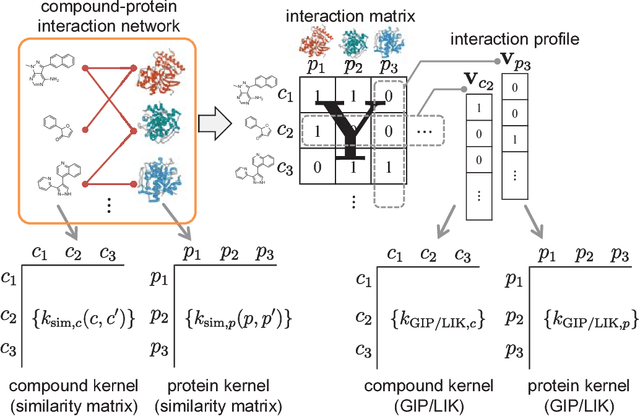
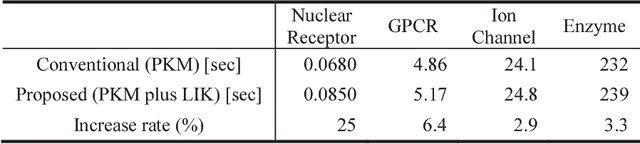
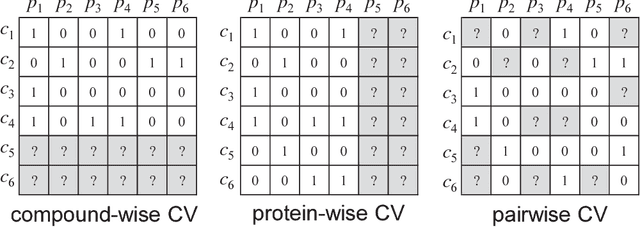
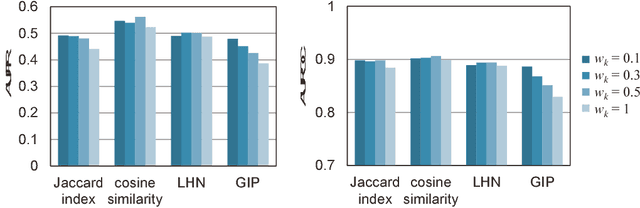
Virtual screening (VS) is widely used during computational drug discovery to reduce costs. Chemogenomics-based virtual screening (CGBVS) can be used to predict new compound-protein interactions (CPIs) from known CPI network data using several methods, including machine learning and data mining. Although CGBVS facilitates highly efficient and accurate CPI prediction, it has poor performance for prediction of new compounds for which CPIs are unknown. The pairwise kernel method (PKM) is a state-of-the-art CGBVS method and shows high accuracy for prediction of new compounds. In this study, on the basis of link mining, we improved the PKM by combining link indicator kernel (LIK) and chemical similarity and evaluated the accuracy of these methods. The proposed method obtained an average area under the precision-recall curve (AUPR) value of 0.562, which was higher than that achieved by the conventional Gaussian interaction profile (GIP) method (0.425), and the calculation time was only increased by a few percent.