 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeXL-Sum: Large-Scale Multilingual Abstractive Summarization for 44 Languages
Jun 25, 2021
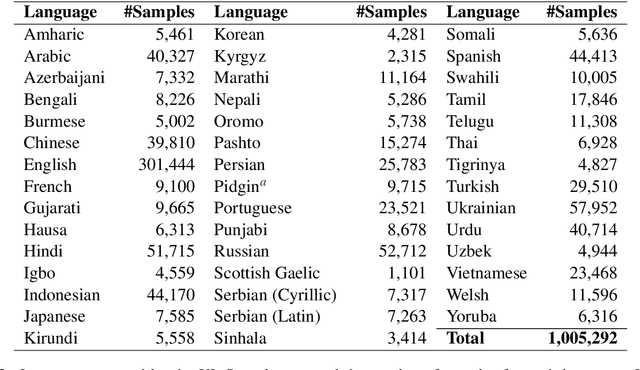
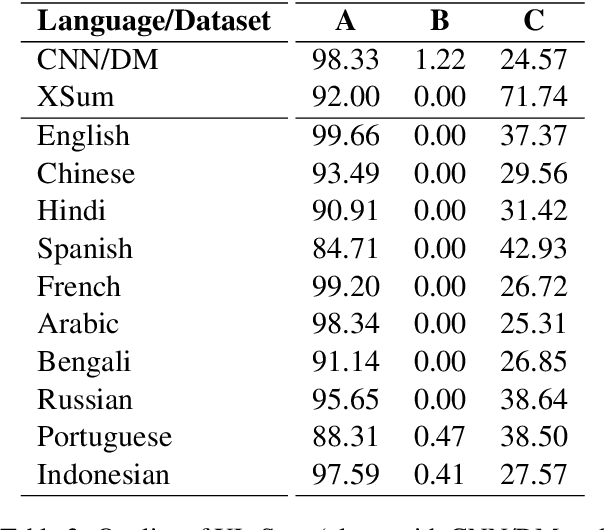
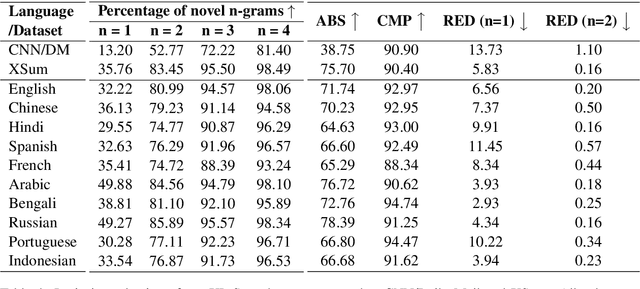
Contemporary works on abstractive text summarization have focused primarily on high-resource languages like English, mostly due to the limited availability of datasets for low/mid-resource ones. In this work, we present XL-Sum, a comprehensive and diverse dataset comprising 1 million professionally annotated article-summary pairs from BBC, extracted using a set of carefully designed heuristics. The dataset covers 44 languages ranging from low to high-resource, for many of which no public dataset is currently available. XL-Sum is highly abstractive, concise, and of high quality, as indicated by human and intrinsic evaluation. We fine-tune mT5, a state-of-the-art pretrained multilingual model, with XL-Sum and experiment on multilingual and low-resource summarization tasks. XL-Sum induces competitive results compared to the ones obtained using similar monolingual datasets: we show higher than 11 ROUGE-2 scores on 10 languages we benchmark on, with some of them exceeding 15, as obtained by multilingual training. Additionally, training on low-resource languages individually also provides competitive performance. To the best of our knowledge, XL-Sum is the largest abstractive summarization dataset in terms of the number of samples collected from a single source and the number of languages covered. We are releasing our dataset and models to encourage future research on multilingual abstractive summarization. The resources can be found at \url{https://github.com/csebuetnlp/xl-sum}.
EDITH :ECG biometrics aided by Deep learning for reliable Individual auTHentication
Feb 16, 2021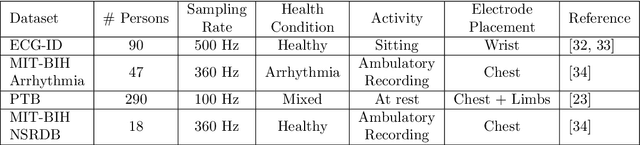
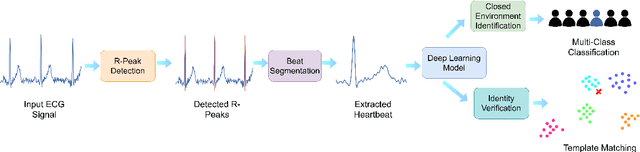
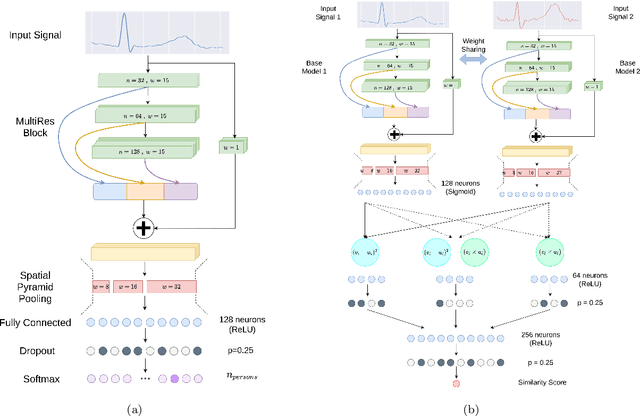
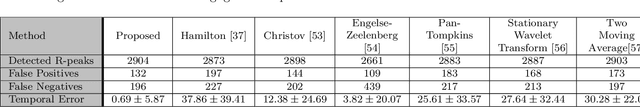
In recent years, physiological signal based authentication has shown great promises,for its inherent robustness against forgery. Electrocardiogram (ECG) signal, being the most widely studied biosignal, has also received the highest level of attention in this regard. It has been proven with numerous studies that by analyzing ECG signals from different persons, it is possible to identify them, with acceptable accuracy. In this work, we present, EDITH, a deep learning-based framework for ECG biometrics authentication system. Moreover, we hypothesize and demonstrate that Siamese architectures can be used over typical distance metrics for improved performance. We have evaluated EDITH using 4 commonly used datasets and outperformed the prior works using less number of beats. EDITH performs competitively using just a single heartbeat (96-99.75% accuracy) and can be further enhanced by fusing multiple beats (100% accuracy from 3 to 6 beats). Furthermore, the proposed Siamese architecture manages to reduce the identity verification Equal Error Rate (EER) to 1.29%. A limited case study of EDITH with real-world experimental data also suggests its potential as a practical authentication system.
BanglaBERT: Combating Embedding Barrier for Low-Resource Language Understanding
Jan 01, 2021Pre-training language models on large volume of data with self-supervised objectives has become a standard practice in natural language processing. However, most such state-of-the-art models are available in only English and other resource-rich languages. Even in multilingual models, which are trained on hundreds of languages, low-resource ones still remain underrepresented. Bangla, the seventh most widely spoken language in the world, is still low in terms of resources. Few downstream task datasets for language understanding in Bangla are publicly available, and there is a clear shortage of good quality data for pre-training. In this work, we build a Bangla natural language understanding model pre-trained on 18.6 GB data we crawled from top Bangla sites on the internet. We introduce a new downstream task dataset and benchmark on four tasks on sentence classification, document classification, natural language understanding, and sequence tagging. Our model outperforms multilingual baselines and previous state-of-the-art results by 1-6%. In the process, we identify a major shortcoming of multilingual models that hurt performance for low-resource languages that don't share writing scripts with any high resource one, which we name the `Embedding Barrier'. We perform extensive experiments to study this barrier. We release all our datasets and pre-trained models to aid future NLP research on Bangla and other low-resource languages. Our code and data are available at https://github.com/csebuetnlp/banglabert.
Align-gram : Rethinking the Skip-gram Model for Protein Sequence Analysis
Dec 06, 2020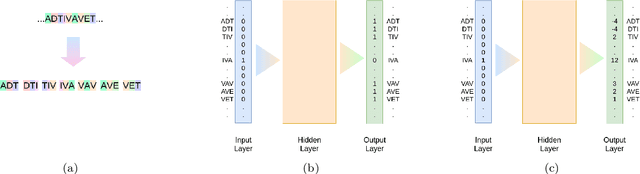
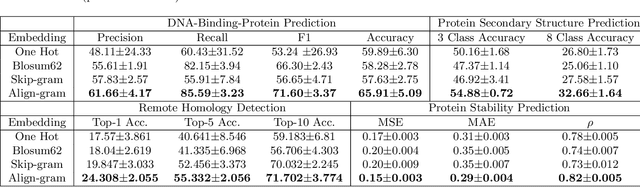
Background: The inception of next generations sequencing technologies have exponentially increased the volume of biological sequence data. Protein sequences, being quoted as the `language of life', has been analyzed for a multitude of applications and inferences. Motivation: Owing to the rapid development of deep learning, in recent years there have been a number of breakthroughs in the domain of Natural Language Processing. Since these methods are capable of performing different tasks when trained with a sufficient amount of data, off-the-shelf models are used to perform various biological applications. In this study, we investigated the applicability of the popular Skip-gram model for protein sequence analysis and made an attempt to incorporate some biological insights into it. Results: We propose a novel $k$-mer embedding scheme, Align-gram, which is capable of mapping the similar $k$-mers close to each other in a vector space. Furthermore, we experiment with other sequence-based protein representations and observe that the embeddings derived from Align-gram aids modeling and training deep learning models better. Our experiments with a simple baseline LSTM model and a much complex CNN model of DeepGoPlus shows the potential of Align-gram in performing different types of deep learning applications for protein sequence analysis.
Not Low-Resource Anymore: Aligner Ensembling, Batch Filtering, and New Datasets for Bengali-English Machine Translation
Oct 07, 2020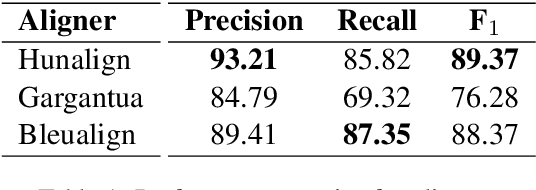
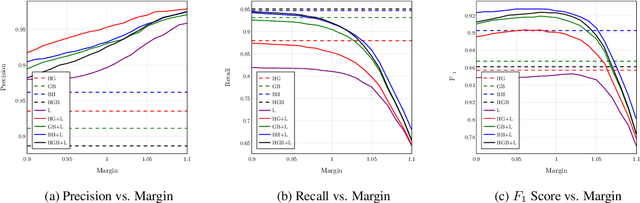
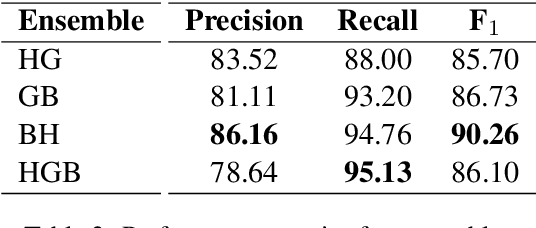
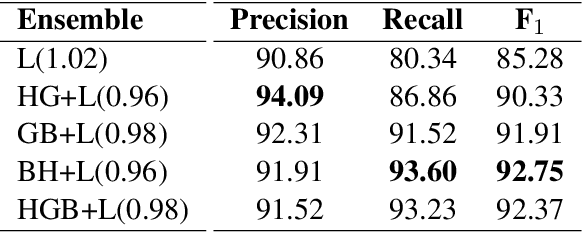
Despite being the seventh most widely spoken language in the world, Bengali has received much less attention in machine translation literature due to being low in resources. Most publicly available parallel corpora for Bengali are not large enough; and have rather poor quality, mostly because of incorrect sentence alignments resulting from erroneous sentence segmentation, and also because of a high volume of noise present in them. In this work, we build a customized sentence segmenter for Bengali and propose two novel methods for parallel corpus creation on low-resource setups: aligner ensembling and batch filtering. With the segmenter and the two methods combined, we compile a high-quality Bengali-English parallel corpus comprising of 2.75 million sentence pairs, more than 2 million of which were not available before. Training on neural models, we achieve an improvement of more than 9 BLEU score over previous approaches to Bengali-English machine translation. We also evaluate on a new test set of 1000 pairs made with extensive quality control. We release the segmenter, parallel corpus, and the evaluation set, thus elevating Bengali from its low-resource status. To the best of our knowledge, this is the first ever large scale study on Bengali-English machine translation. We believe our study will pave the way for future research on Bengali-English machine translation as well as other low-resource languages. Our data and code are available at https://github.com/csebuetnlp/banglanmt.
COVID-19Base: A knowledgebase to explore biomedical entities related to COVID-19
May 12, 2020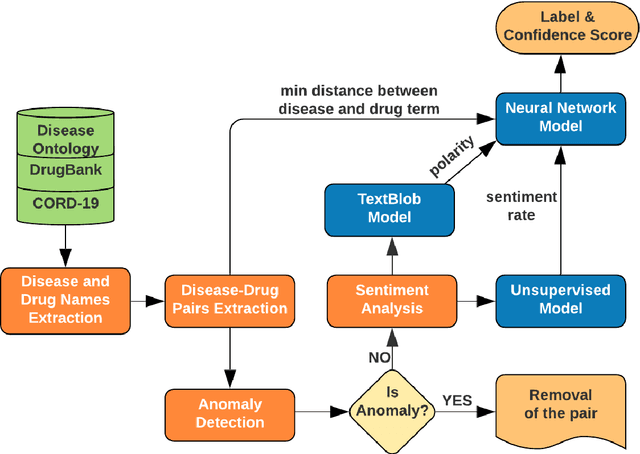
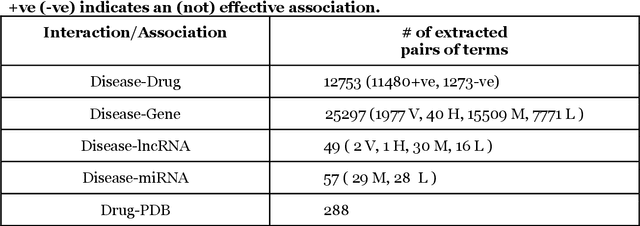
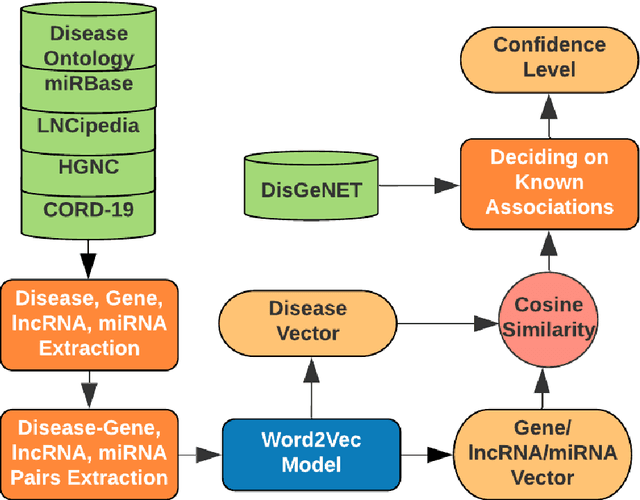
We are presenting COVID-19Base, a knowledgebase highlighting the biomedical entities related to COVID-19 disease based on literature mining. To develop COVID-19Base, we mine the information from publicly available scientific literature and related public resources. We considered seven topic-specific dictionaries, including human genes, human miRNAs, human lncRNAs, diseases, Protein Databank, drugs, and drug side effects, are integrated to mine all scientific evidence related to COVID-19. We have employed an automated literature mining and labeling system through a novel approach to measure the effectiveness of drugs against diseases based on natural language processing, sentiment analysis, and deep learning. To the best of our knowledge, this is the first knowledgebase dedicated to COVID-19, which integrates such large variety of related biomedical entities through literature mining. Proper investigation of the mined biomedical entities along with the identified interactions among those, reported in COVID-19Base, would help the research community to discover possible ways for the therapeutic treatment of COVID-19.
PPG2ABP: Translating Photoplethysmogram (PPG) Signals to Arterial Blood Pressure (ABP) Waveforms using Fully Convolutional Neural Networks
May 04, 2020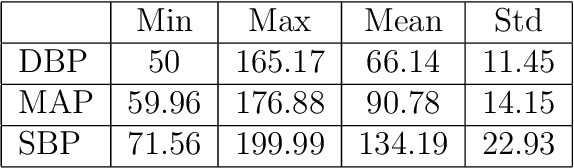

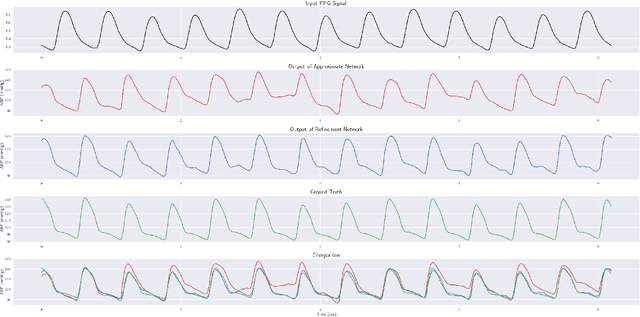
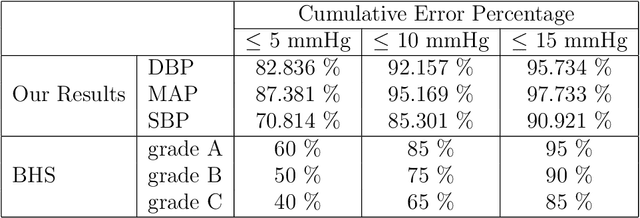
Cardiovascular diseases are one of the most severe causes of mortality, taking a heavy toll of lives annually throughout the world. The continuous monitoring of blood pressure seems to be the most viable option, but this demands an invasive process, bringing about several layers of complexities. This motivates us to develop a method to predict the continuous arterial blood pressure (ABP) waveform through a non-invasive approach using photoplethysmogram (PPG) signals. In addition we explore the advantage of deep learning as it would free us from sticking to ideally shaped PPG signals only, by making handcrafted feature computation irrelevant, which is a shortcoming of the existing approaches. Thus, we present, PPG2ABP, a deep learning based method, that manages to predict the continuous ABP waveform from the input PPG signal, with a mean absolute error of 4.604 mmHg, preserving the shape, magnitude and phase in unison. However, the more astounding success of PPG2ABP turns out to be that the computed values of DBP, MAP and SBP from the predicted ABP waveform outperforms the existing works under several metrics, despite that PPG2ABP is not explicitly trained to do so.
MultiResUNet : Rethinking the U-Net Architecture for Multimodal Biomedical Image Segmentation
Feb 11, 2019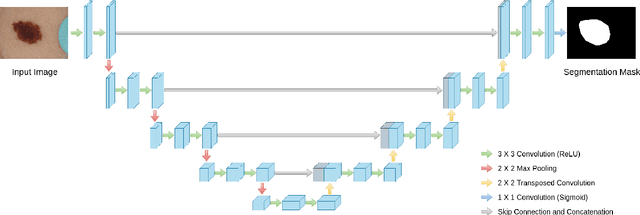
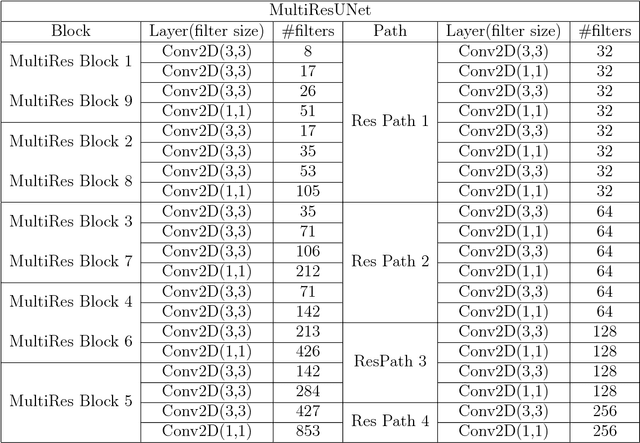
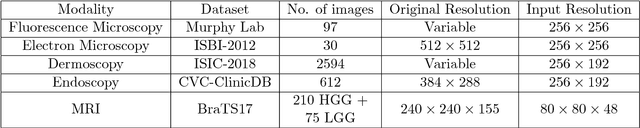
In recent years Deep Learning has brought about a breakthrough in Medical Image Segmentation. U-Net is the most prominent deep network in this regard, which has been the most popular architecture in the medical imaging community. Despite outstanding overall performance in segmenting multimodal medical images, from extensive experimentations on challenging datasets, we found out that the classical U-Net architecture seems to be lacking in certain aspects. Therefore, we propose some modifications to improve upon the already state-of-the-art U-Net model. Hence, following the modifications we develop a novel architecture MultiResUNet as the potential successor to the successful U-Net architecture. We have compared our proposed architecture MultiResUNet with the classical U-Net on a vast repertoire of multimodal medical images. Albeit slight improvements in the cases of ideal images, a remarkable gain in performance has been attained for challenging images. We have evaluated our model on five different datasets, each with their own unique challenges, and have obtained a relative improvement in performance of 10.15%, 5.07%, 2.63%, 1.41%, and 0.62% respectively.
Deep Face Image Retrieval: a Comparative Study with Dictionary Learning
Dec 13, 2018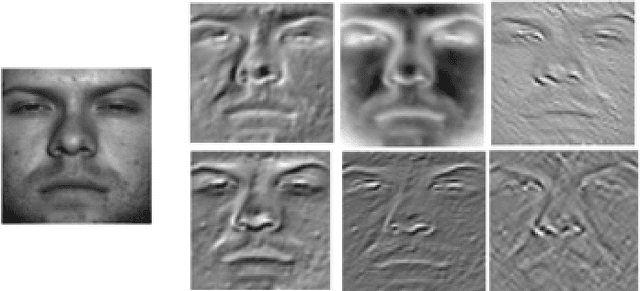
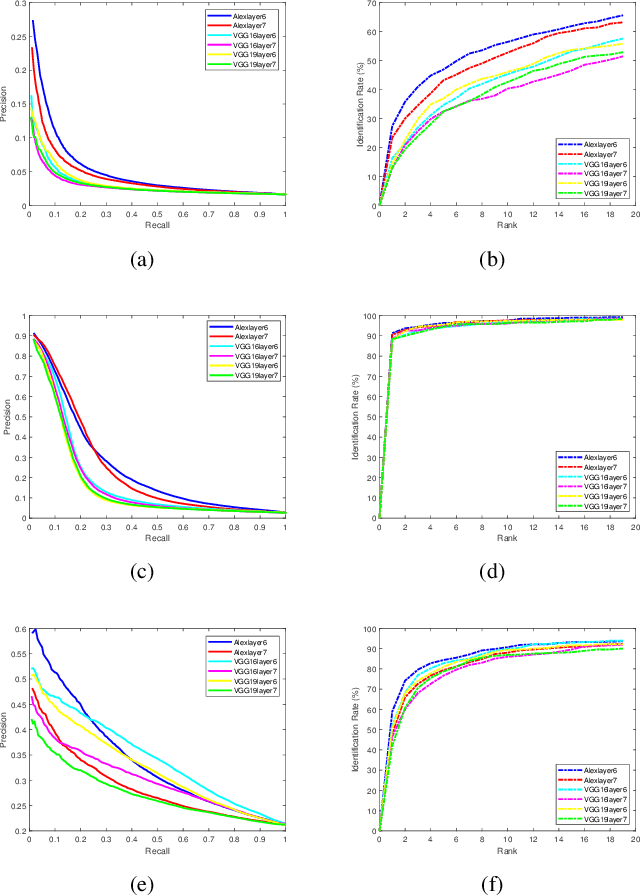
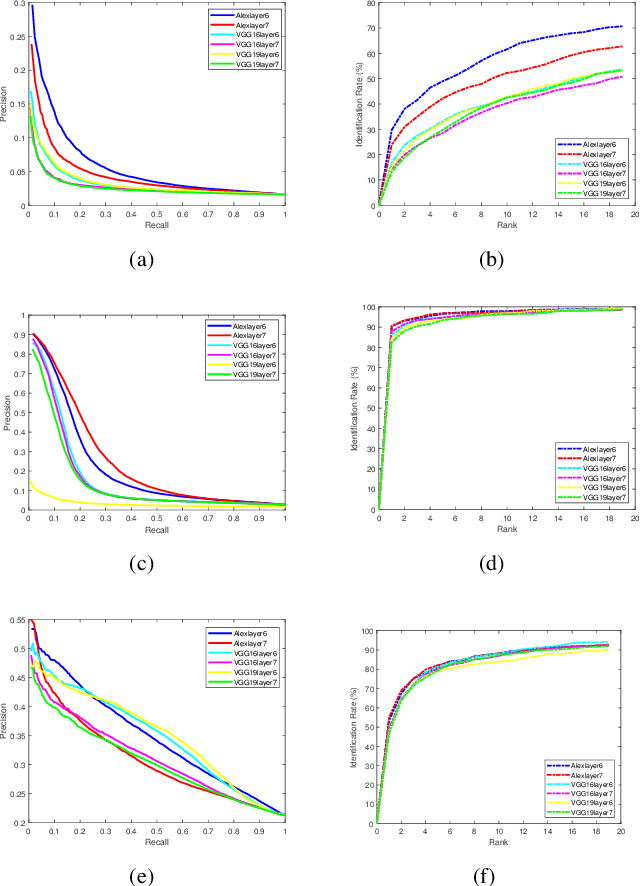
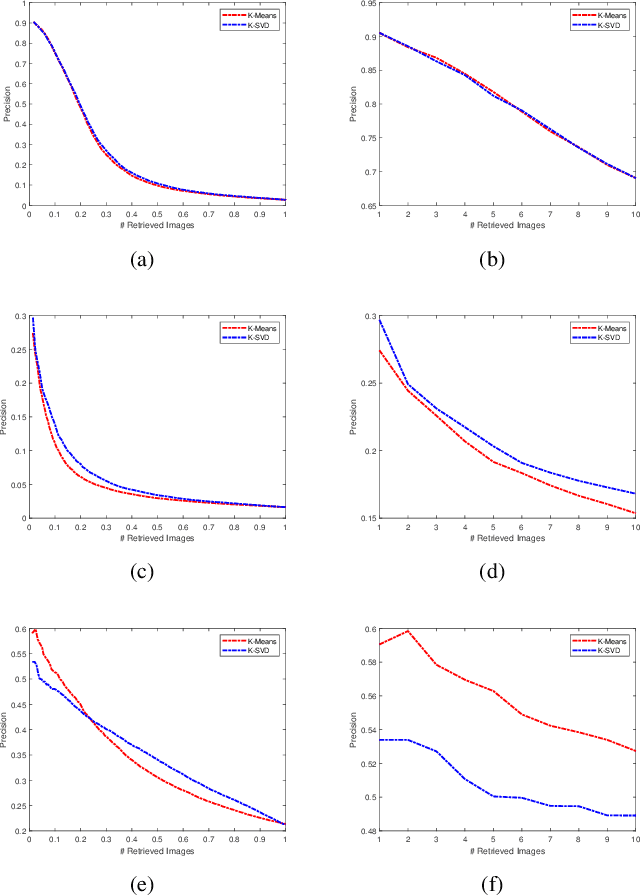
Facial image retrieval is a challenging task since faces have many similar features (areas), which makes it difficult for the retrieval systems to distinguish faces of different people. With the advent of deep learning, deep networks are often applied to extract powerful features that are used in many areas of computer vision. This paper investigates the application of different deep learning models for face image retrieval, namely, Alexlayer6, Alexlayer7, VGG16layer6, VGG16layer7, VGG19layer6, and VGG19layer7, with two types of dictionary learning techniques, namely $K$-means and $K$-SVD. We also investigate some coefficient learning techniques such as the Homotopy, Lasso, Elastic Net and SSF and their effect on the face retrieval system. The comparative results of the experiments conducted on three standard face image datasets show that the best performers for face image retrieval are Alexlayer7 with $K$-means and SSF, Alexlayer6 with $K$-SVD and SSF, and Alexlayer6 with $K$-means and SSF. The APR and ARR of these methods were further compared to some of the state of the art methods based on local descriptors. The experimental results show that deep learning outperforms most of those methods and therefore can be recommended for use in practice of face image retrieval
VFPred: A Fusion of Signal Processing and Machine Learning techniques in Detecting Ventricular Fibrillation from ECG Signals
Jul 24, 2018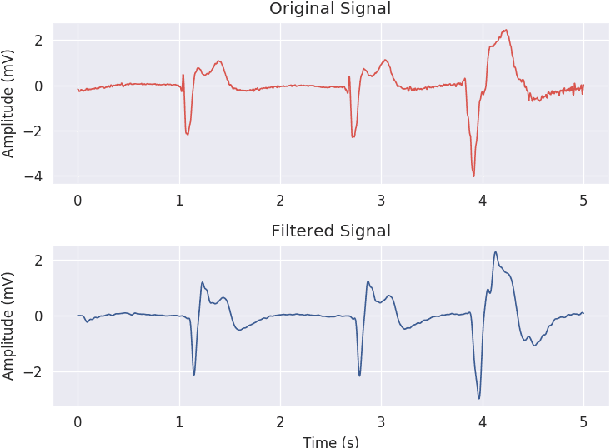
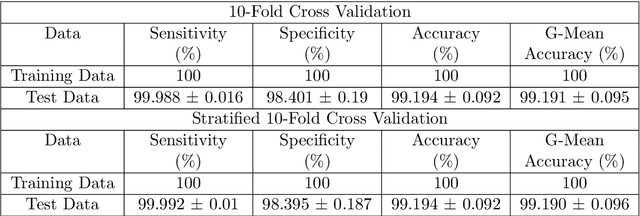
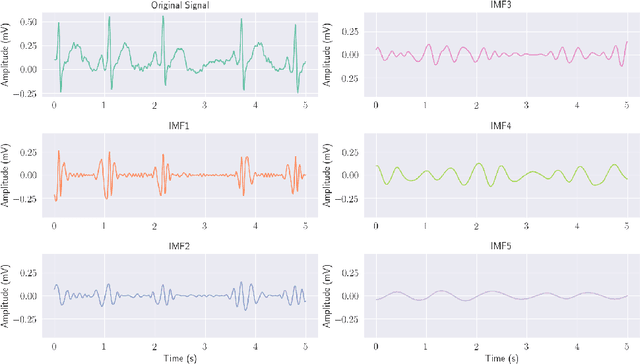
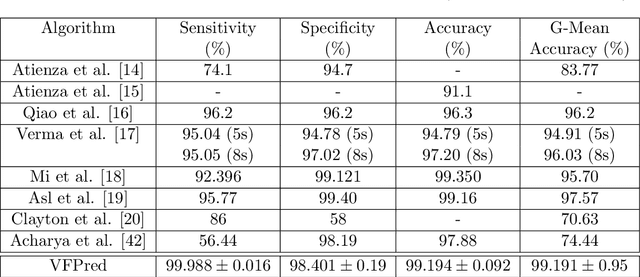
Ventricular Fibrillation (VF), one of the most dangerous arrhythmias, is responsible for sudden cardiac arrests. Thus, various algorithms have been developed to predict VF from Electrocardiogram (ECG), which is a binary classification problem. In the literature, we find a number of algorithms based on signal processing, where, after some robust mathematical operations the decision is given based on a predefined threshold over a single value. On the other hand, some machine learning based algorithms are also reported in the literature; however, these algorithms merely combine some parameters and make a prediction using those as features. Both the approaches have their perks and pitfalls; thus our motivation was to coalesce them to get the best out of the both worlds. Hence we have developed, VFPred that, in addition to employing a signal processing pipeline, namely, Empirical Mode Decomposition and Discrete Time Fourier Transform for useful feature extraction, uses a Support Vector Machine for efficient classification. VFPred turns out to be a robust algorithm as it is able to successfully segregate the two classes with equal confidence (Sensitivity = 99.99%, Specificity = 98.40%) even from a short signal of 5 seconds long, whereas existing works though requires longer signals, flourishes in one but fails in the other.