 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA 96pJ/Frame/Pixel and 61pJ/Event Anti-UAV System with Hybrid Object Tracking Modes
Dec 12, 2025We present an energy-efficient anti-UAV system that integrates frame-based and event-driven object tracking to enable reliable detection of small and fast-moving drones. The system reconstructs binary event frames using run-length encoding, generates region proposals, and adaptively switches between frame mode and event mode based on object size and velocity. A Fast Object Tracking Unit improves robustness for high-speed targets through adaptive thresholding and trajectory-based classification. The neural processing unit supports both grayscale-patch and trajectory inference with a custom instruction set and a zero-skipping MAC architecture, reducing redundant neural computations by more than 97 percent. Implemented in 40 nm CMOS technology, the 2 mm^2 chip achieves 96 pJ per frame per pixel and 61 pJ per event at 0.8 V, and reaches 98.2 percent recognition accuracy on public UAV datasets across 50 to 400 m ranges and 5 to 80 pixels per second speeds. The results demonstrate state-of-the-art end-to-end energy efficiency for anti-UAV systems.
Deep Rotation Equivariant Network
Feb 28, 2018



Recently, learning equivariant representations has attracted considerable research attention. Dieleman et al. introduce four operations which can be inserted into convolutional neural network to learn deep representations equivariant to rotation. However, feature maps should be copied and rotated four times in each layer in their approach, which causes much running time and memory overhead. In order to address this problem, we propose Deep Rotation Equivariant Network consisting of cycle layers, isotonic layers and decycle layers. Our proposed layers apply rotation transformation on filters rather than feature maps, achieving a speed up of more than 2 times with even less memory overhead. We evaluate DRENs on Rotated MNIST and CIFAR-10 datasets and demonstrate that it can improve the performance of state-of-the-art architectures.
Learning Graph-Level Representation for Drug Discovery
Sep 16, 2017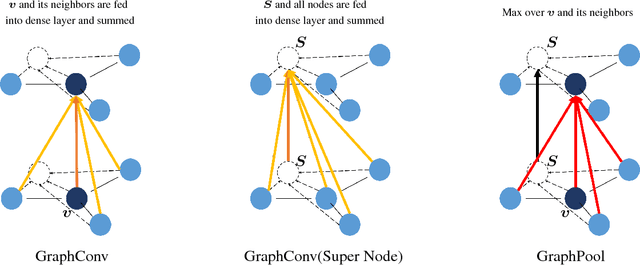
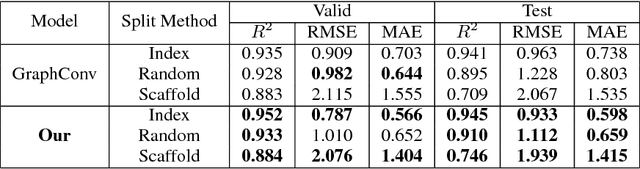
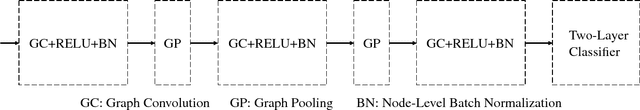
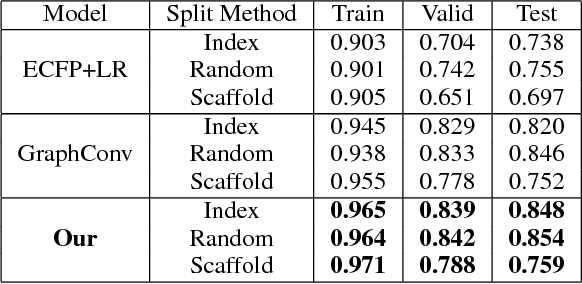
Predicating macroscopic influences of drugs on human body, like efficacy and toxicity, is a central problem of small-molecule based drug discovery. Molecules can be represented as an undirected graph, and we can utilize graph convolution networks to predication molecular properties. However, graph convolutional networks and other graph neural networks all focus on learning node-level representation rather than graph-level representation. Previous works simply sum all feature vectors for all nodes in the graph to obtain the graph feature vector for drug predication. In this paper, we introduce a dummy super node that is connected with all nodes in the graph by a directed edge as the representation of the graph and modify the graph operation to help the dummy super node learn graph-level feature. Thus, we can handle graph-level classification and regression in the same way as node-level classification and regression. In addition, we apply focal loss to address class imbalance in drug datasets. The experiments on MoleculeNet show that our method can effectively improve the performance of molecular properties predication.