 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePerspective: Towards sustainable exploration of chemical spaces with machine learning
Mar 31, 2026Artificial intelligence is transforming molecular and materials science, but its growing computational and data demands raise critical sustainability challenges. In this Perspective, we examine resource considerations across the AI-driven discovery pipeline--from quantum-mechanical (QM) data generation and model training to automated, self-driving research workflows--building on discussions from the ``SusML workshop: Towards sustainable exploration of chemical spaces with machine learning'' held in Dresden, Germany. In this context, the availability of large quantum datasets has enabled rigorous benchmarking and rapid methodological progress, while also incurring substantial energy and infrastructure costs. We highlight emerging strategies to enhance efficiency, including general-purpose machine learning (ML) models, multi-fidelity approaches, model distillation, and active learning. Moreover, incorporating physics-based constraints within hierarchical workflows, where fast ML surrogates are applied broadly and high-accuracy QM methods are used selectively, can further optimize resource use without compromising reliability. Equally important is bridging the gap between idealized computational predictions and real-world conditions by accounting for synthesizability and multi-objective design criteria, which is essential for practical impact. Finally, we argue that sustainable progress will rely on open data and models, reusable workflows, and domain-specific AI systems that maximize scientific value per unit of computation, enabling efficient and responsible discovery of technological materials and therapeutics.
Design Space of Self--Consistent Electrostatic Machine Learning Interatomic Potentials
Mar 16, 2026Machine learning interatomic potentials (MLIPs) have become widely used tools in atomistic simulations. For much of the history of this field, the most commonly employed architectures were based on short-ranged atomic energy contributions, and the assumption of locality still persists in many modern foundation models. While this approach has enabled efficient and accurate modelling for many use cases, it poses intrinsic limitations for systems where long-range electrostatics, charge transfer, or induced polarization play a central role. A growing body of work has proposed extensions that incorporate electrostatic effects, ranging from locally predicted atomic charges to self-consistent models. While these models have demonstrated success for specific examples, their underlying assumptions, and fundamental limitations are not yet well understood. In this work, we present a framework for treating electrostatics in MLIPs by viewing existing models as coarse-grained approximations to density functional theory (DFT). This perspective makes explicit the approximations involved, clarifies the physical meaning of the learned quantities, and reveals connections and equivalences between several previously proposed models. Using this formalism, we identify key design choices that define a broader design space of self-consistent electrostatic MLIPs. We implement salient points in this space using the MACE architecture and a shared representation of the charge density, enabling controlled comparisons between different approaches. Finally, we evaluate these models on two instructive test cases: metal-water interfaces, which probe the contrasting electrostatic response of conducting and insulating systems, and charged vacancies in silicon dioxide. Our results highlight the limitations of existing approaches and demonstrate how more expressive self-consistent models are needed to resolve failures.
Fast and Uncertainty-Aware Directional Message Passing for Non-Equilibrium Molecules
Dec 01, 2020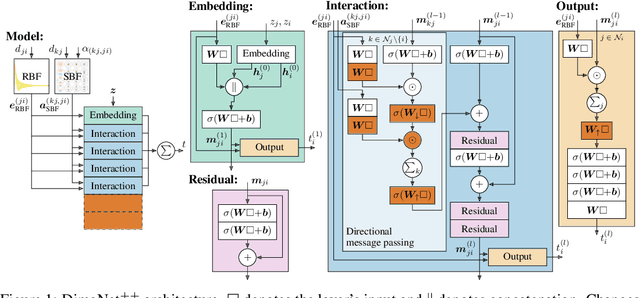

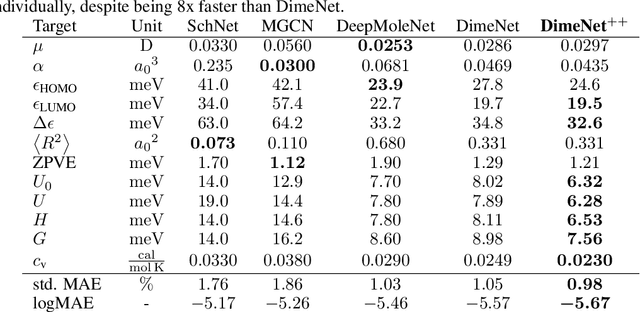
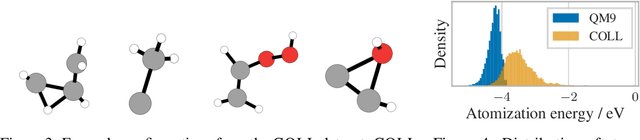
Many important tasks in chemistry revolve around molecules during reactions. This requires predictions far from the equilibrium, while most recent work in machine learning for molecules has been focused on equilibrium or near-equilibrium states. In this paper we aim to extend this scope in three ways. First, we propose the DimeNet++ model, which is 8x faster and 10% more accurate than the original DimeNet on the QM9 benchmark of equilibrium molecules. Second, we validate DimeNet++ on highly reactive molecules by developing the challenging COLL dataset, which contains distorted configurations of small molecules during collisions. Finally, we investigate ensembling and mean-variance estimation for uncertainty quantification with the goal of accelerating the exploration of the vast space of non-equilibrium structures. Our DimeNet++ implementation as well as the COLL dataset are available online.