 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeComposition based oxidation state prediction of materials using deep learning
Nov 29, 2022



Oxidation states are the charges of atoms after their ionic approximation of their bonds, which have been widely used in charge-neutrality verification, crystal structure determination, and reaction estimation. Currently only heuristic rules exist for guessing the oxidation states of a given compound with many exceptions. Recent work has developed machine learning models based on heuristic structural features for predicting the oxidation states of metal ions. However, composition based oxidation state prediction still remains elusive so far, which is more important in new material discovery for which the structures are not even available. This work proposes a novel deep learning based BERT transformer language model BERTOS for predicting the oxidation states of all elements of inorganic compounds given only their chemical composition. Our model achieves 96.82\% accuracy for all-element oxidation states prediction benchmarked on the cleaned ICSD dataset and achieves 97.61\% accuracy for oxide materials. We also demonstrate how it can be used to conduct large-scale screening of hypothetical material compositions for materials discovery.
Materials Property Prediction with Uncertainty Quantification: A Benchmark Study
Nov 11, 2022



Uncertainty quantification (UQ) has increasing importance in building robust high-performance and generalizable materials property prediction models. It can also be used in active learning to train better models by focusing on getting new training data from uncertain regions. There are several categories of UQ methods each considering different types of uncertainty sources. Here we conduct a comprehensive evaluation on the UQ methods for graph neural network based materials property prediction and evaluate how they truly reflect the uncertainty that we want in error bound estimation or active learning. Our experimental results over four crystal materials datasets (including formation energy, adsorption energy, total energy, and band gap properties) show that the popular ensemble methods for uncertainty estimation is NOT the best choice for UQ in materials property prediction. For the convenience of the community, all the source code and data sets can be accessed freely at \url{https://github.com/usccolumbia/materialsUQ}.
Probabilistic Generative Transformer Language models for Generative Design of Molecules
Sep 20, 2022
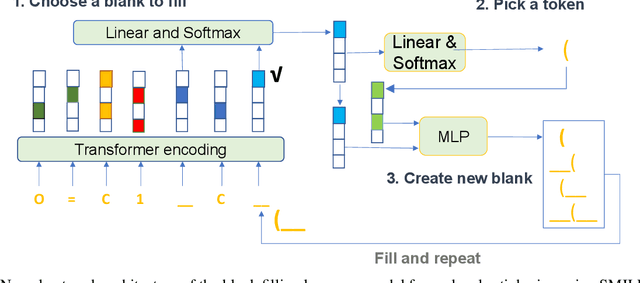
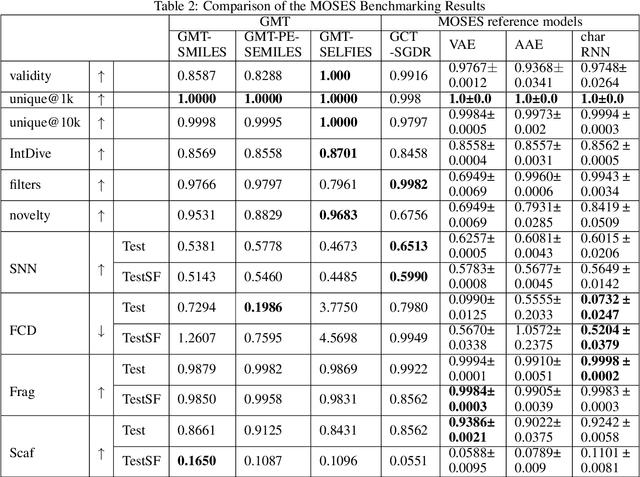
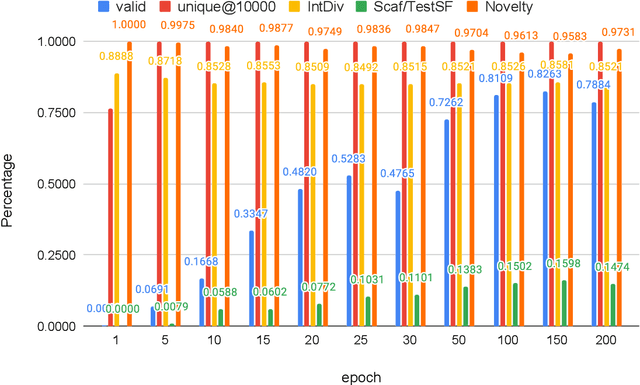
Self-supervised neural language models have recently found wide applications in generative design of organic molecules and protein sequences as well as representation learning for downstream structure classification and functional prediction. However, most of the existing deep learning models for molecule design usually require a big dataset and have a black-box architecture, which makes it difficult to interpret their design logic. Here we propose Generative Molecular Transformer (GMTransformer), a probabilistic neural network model for generative design of molecules. Our model is built on the blank filling language model originally developed for text processing, which has demonstrated unique advantages in learning the "molecules grammars" with high-quality generation, interpretability, and data efficiency. Benchmarked on the MOSES datasets, our models achieve high novelty and Scaf compared to other baselines. The probabilistic generation steps have the potential in tinkering molecule design due to their capability of recommending how to modify existing molecules with explanation, guided by the learned implicit molecule chemistry. The source code and datasets can be accessed freely at https://github.com/usccolumbia/GMTransformer
Materials Transformers Language Models for Generative Materials Design: a benchmark study
Jun 27, 2022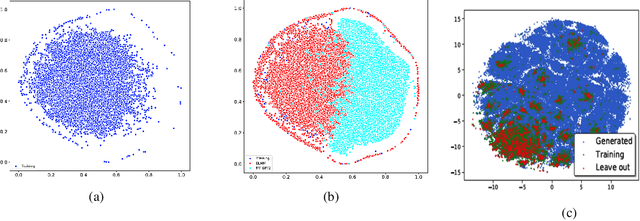
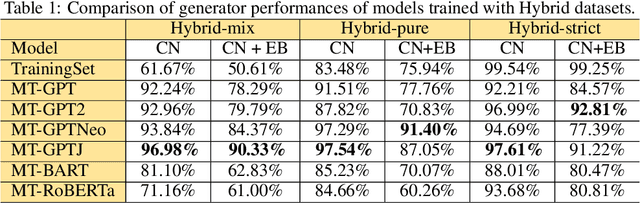
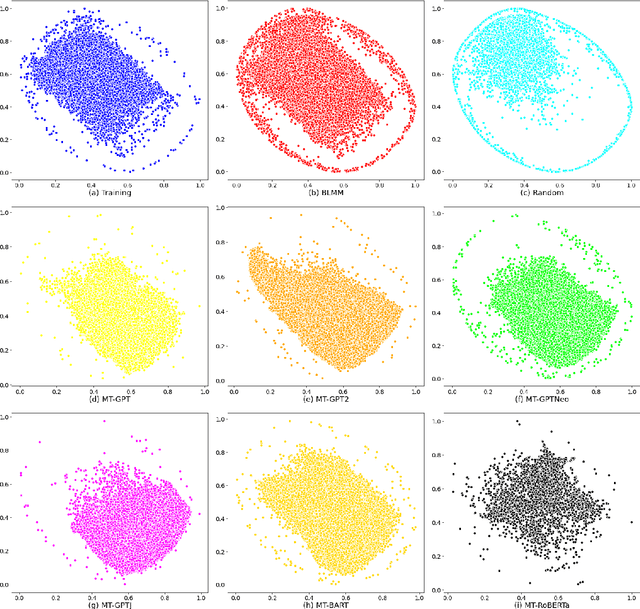
Pre-trained transformer language models on large unlabeled corpus have produced state-of-the-art results in natural language processing, organic molecule design, and protein sequence generation. However, no such models have been applied to learn the composition patterns of inorganic materials. Here we train a series of seven modern transformer language models (GPT, GPT-2, GPT-Neo, GPT-J, BLMM, BART, and RoBERTa) using the expanded formulas from material deposited in the ICSD, OQMD, and Materials Projects databases. Six different datasets with/out non-charge-neutral or balanced electronegativity samples are used to benchmark the performances and uncover the generation biases of modern transformer models for the generative design of materials compositions. Our extensive experiments showed that the causal language models based materials transformers can generate chemically valid materials compositions with as high as 97.54\% to be charge neutral and 91.40\% to be electronegativity balanced, which has more than 6 times higher enrichment compared to a baseline pseudo-random sampling algorithm. These models also demonstrate high novelty and their potential in new materials discovery has been proved by their capability to recover the leave-out materials. We also find that the properties of the generated samples can be tailored by training the models with selected training sets such as high-bandgap materials. Our experiments also showed that different models each have their own preference in terms of the properties of the generated samples and their running time complexity varies a lot. We have applied our materials transformer models to discover a set of new materials as validated using DFT calculations.
Crystal Transformer: Self-learning neural language model for Generative and Tinkering Design of Materials
Apr 25, 2022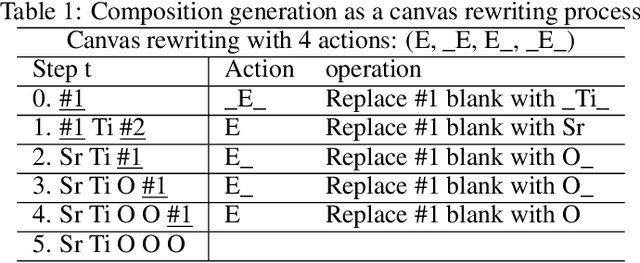
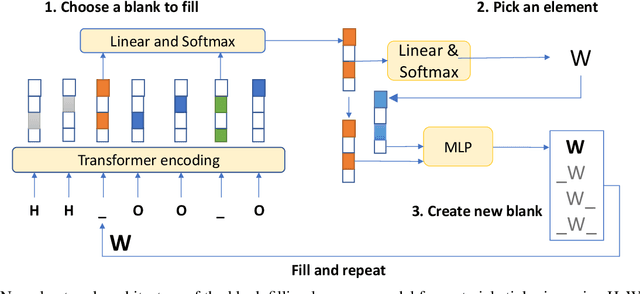
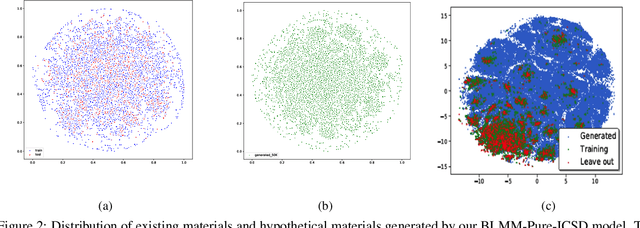
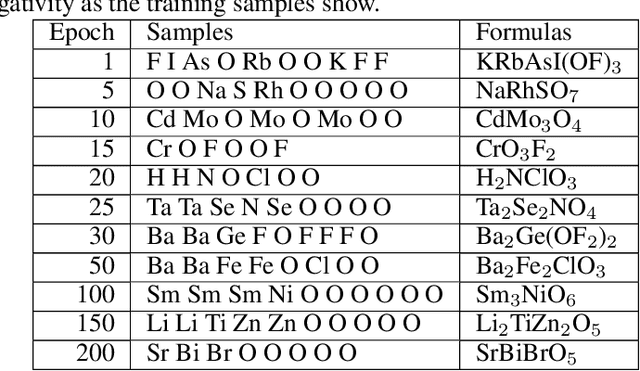
Self-supervised neural language models have recently achieved unprecedented success, from natural language processing to learning the languages of biological sequences and organic molecules. These models have demonstrated superior performance in the generation, structure classification, and functional predictions for proteins and molecules with learned representations. However, most of the masking-based pre-trained language models are not designed for generative design, and their black-box nature makes it difficult to interpret their design logic. Here we propose BLMM Crystal Transformer, a neural network based probabilistic generative model for generative and tinkering design of inorganic materials. Our model is built on the blank filling language model for text generation and has demonstrated unique advantages in learning the "materials grammars" together with high-quality generation, interpretability, and data efficiency. It can generate chemically valid materials compositions with as high as 89.7\% charge neutrality and 84.8\% balanced electronegativity, which are more than 4 and 8 times higher compared to a pseudo random sampling baseline. The probabilistic generation process of BLMM allows it to recommend tinkering operations based on learned materials chemistry and makes it useful for materials doping. Combined with the TCSP crysal structure prediction algorithm, We have applied our model to discover a set of new materials as validated using DFT calculations. Our work thus brings the unsupervised transformer language models based generative artificial intelligence to inorganic materials. A user-friendly web app has been developed for computational materials doping and can be accessed freely at \url{www.materialsatlas.org/blmtinker}.
Physics Guided Generative Adversarial Networks for Generations of Crystal Materials with Symmetry Constraints
Mar 27, 2022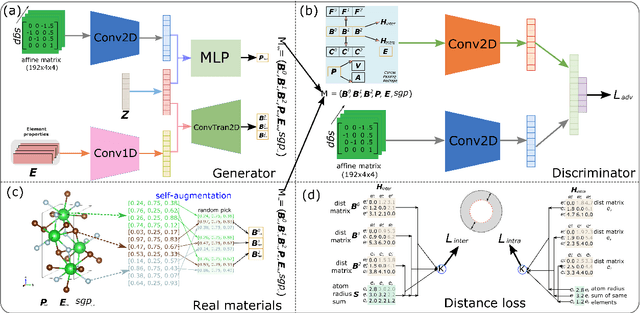
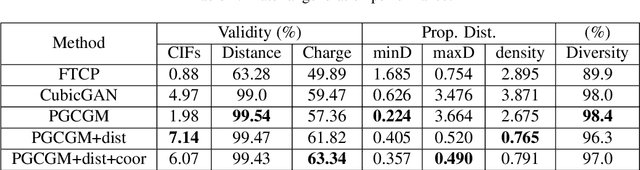

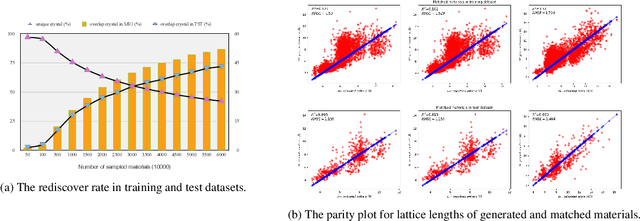
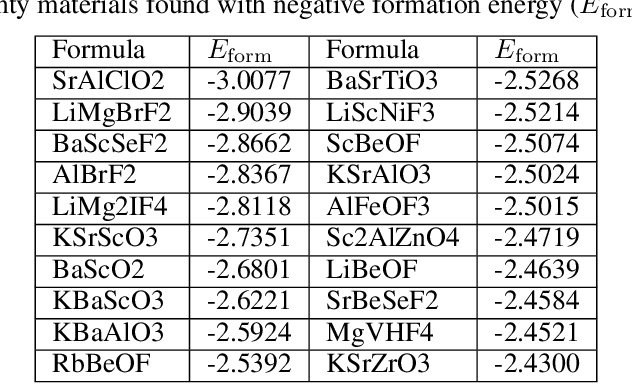
Discovering new materials is a long-standing challenging task that is critical to the progress of human society. Conventional approaches such as trial-and-error experiments and computational simulations are labor-intensive or costly with their success heavily depending on experts' heuristics. Recently deep generative models have been successfully proposed for materials generation by learning implicit knowledge from known materials datasets, with performance however limited by their confinement to a special material family or failing to incorporate physical rules into the model training process. Here we propose a Physics Guided Crystal Generative Model (PGCGM) for new materials generation, which captures and exploits the pairwise atomic distance constraints among neighbor atoms and symmetric geometric constraints. By augmenting the base atom sites of materials, our model can generates new materials of 20 space groups. With atom clustering and merging on generated crystal structures, our method increases the generator's validity by 8 times compared to one of the baselines and by 143\% compared to the previous CubicGAN along with its superiority in properties distribution and diversity. We further validated our generated candidates by Density Functional Theory (DFT) calculation, which successfully optimized/relaxed 1869 materials out of 2000, of which 39.6\% are with negative formation energy, indicating their stability.
Semi-supervised teacher-student deep neural network for materials discovery
Dec 12, 2021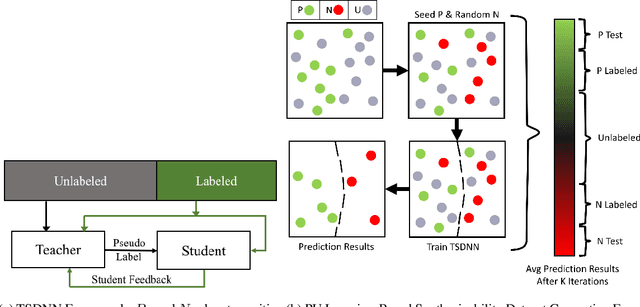
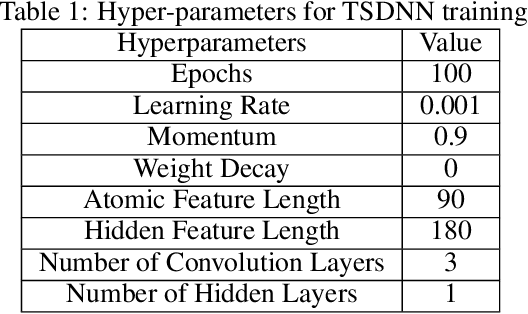
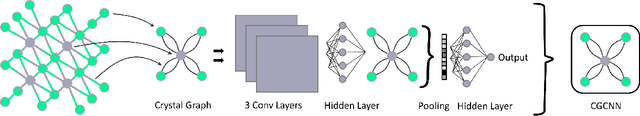
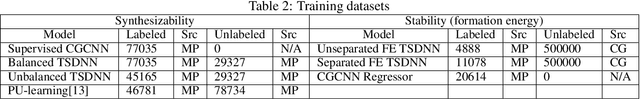
Data driven generative machine learning models have recently emerged as one of the most promising approaches for new materials discovery. While the generator models can generate millions of candidates, it is critical to train fast and accurate machine learning models to filter out stable, synthesizable materials with desired properties. However, such efforts to build supervised regression or classification screening models have been severely hindered by the lack of unstable or unsynthesizable samples, which usually are not collected and deposited in materials databases such as ICSD and Materials Project (MP). At the same time, there are a significant amount of unlabelled data available in these databases. Here we propose a semi-supervised deep neural network (TSDNN) model for high-performance formation energy and synthesizability prediction, which is achieved via its unique teacher-student dual network architecture and its effective exploitation of the large amount of unlabeled data. For formation energy based stability screening, our semi-supervised classifier achieves an absolute 10.3\% accuracy improvement compared to the baseline CGCNN regression model. For synthesizability prediction, our model significantly increases the baseline PU learning's true positive rate from 87.9\% to 97.9\% using 1/49 model parameters. To further prove the effectiveness of our models, we combined our TSDNN-energy and TSDNN-synthesizability models with our CubicGAN generator to discover novel stable cubic structures. Out of 1000 recommended candidate samples by our models, 512 of them have negative formation energies as validated by our DFT formation energy calculations. Our experimental results show that our semi-supervised deep neural networks can significantly improve the screening accuracy in large-scale generative materials design.
Physics guided deep learning generative models for crystal materials discovery
Dec 07, 2021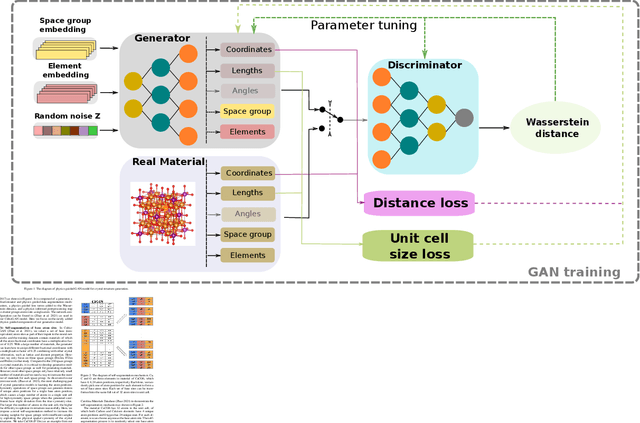
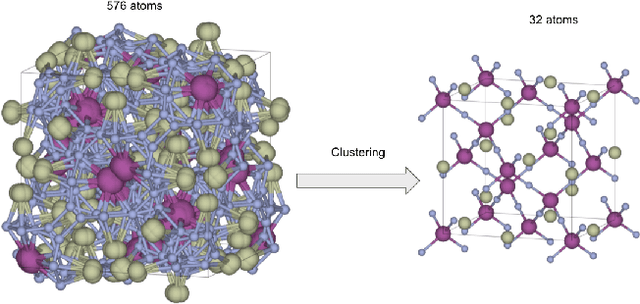
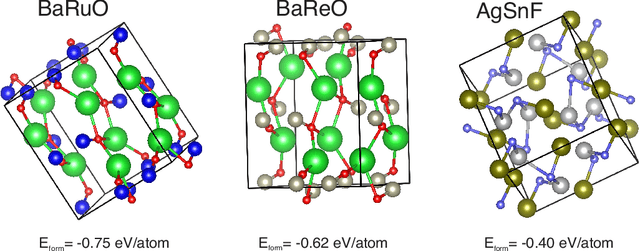
Deep learning based generative models such as deepfake have been able to generate amazing images and videos. However, these models may need significant transformation when applied to generate crystal materials structures in which the building blocks, the physical atoms are very different from the pixels. Naively transferred generative models tend to generate a large portion of physically infeasible crystal structures that are not stable or synthesizable. Herein we show that by exploiting and adding physically oriented data augmentation, loss function terms, and post processing, our deep adversarial network (GAN) based generative models can now generate crystal structures with higher physical feasibility and expand our previous models which can only create cubic structures.
Predicting Lattice Phonon Vibrational Frequencies Using Deep Graph Neural Networks
Nov 10, 2021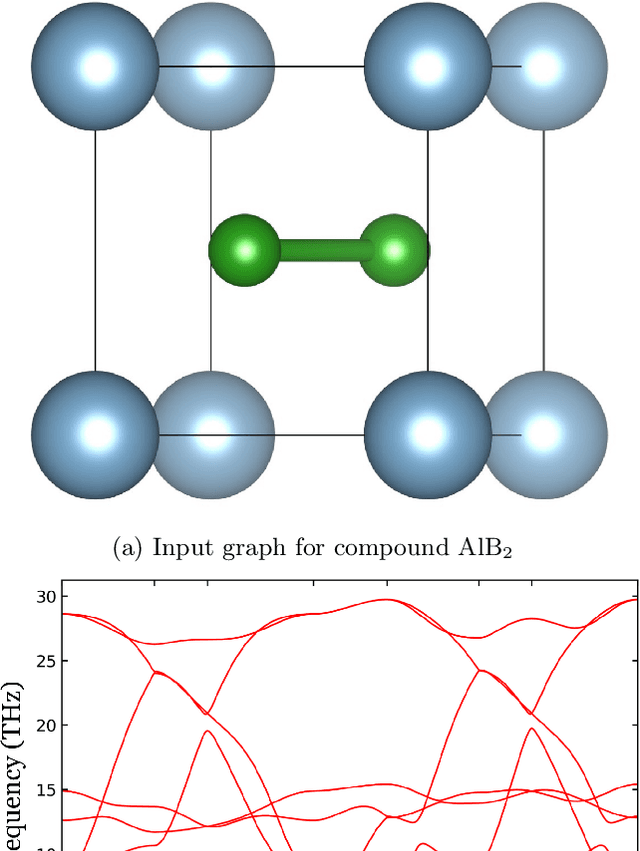
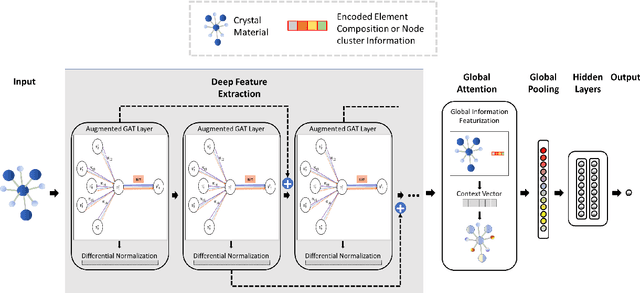
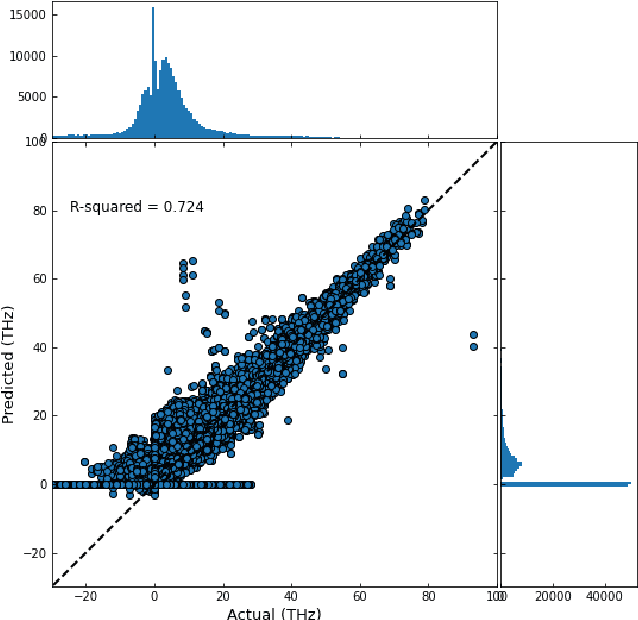
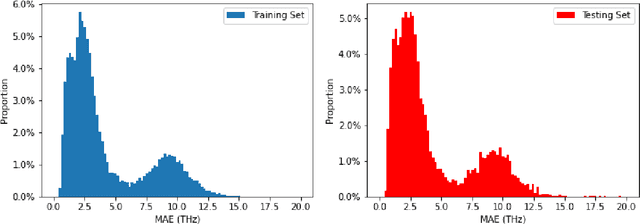
Lattice vibration frequencies are related to many important materials properties such as thermal and electrical conductivity as well as superconductivity. However, computational calculation of vibration frequencies using density functional theory (DFT) methods is too computationally demanding for a large number of samples in materials screening. Here we propose a deep graph neural network-based algorithm for predicting crystal vibration frequencies from crystal structures with high accuracy. Our algorithm addresses the variable dimension of vibration frequency spectrum using the zero padding scheme. Benchmark studies on two data sets with 15,000 and 35,552 samples show that the aggregated $R^2$ scores of the prediction reaches 0.554 and 0.724 respectively. Our work demonstrates the capability of deep graph neural networks to learn to predict phonon spectrum properties of crystal structures in addition to phonon density of states (DOS) and electronic DOS in which the output dimension is constant.
Scalable deeper graph neural networks for high-performance materials property prediction
Sep 25, 2021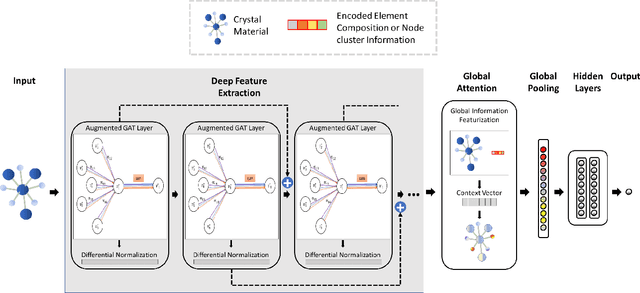
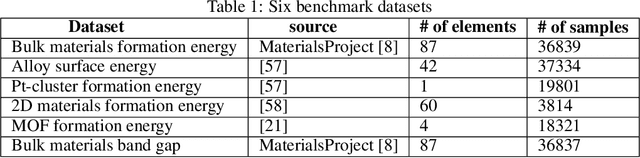
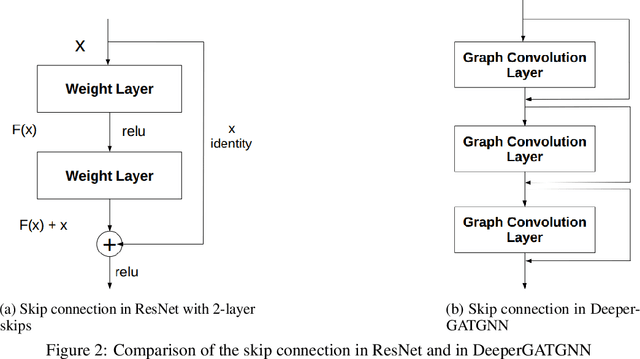
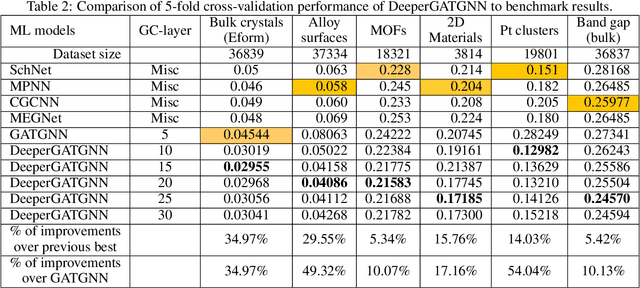
Machine learning (ML) based materials discovery has emerged as one of the most promising approaches for breakthroughs in materials science. While heuristic knowledge based descriptors have been combined with ML algorithms to achieve good performance, the complexity of the physicochemical mechanisms makes it urgently needed to exploit representation learning from either compositions or structures for building highly effective materials machine learning models. Among these methods, the graph neural networks have shown the best performance by its capability to learn high-level features from crystal structures. However, all these models suffer from their inability to scale up the models due to the over-smoothing issue of their message-passing GNN architecture. Here we propose a novel graph attention neural network model DeeperGATGNN with differentiable group normalization and skip-connections, which allows to train very deep graph neural network models (e.g. 30 layers compared to 3-9 layers in previous works). Through systematic benchmark studies over six benchmark datasets for energy and band gap predictions, we show that our scalable DeeperGATGNN model needs little costly hyper-parameter tuning for different datasets and achieves the state-of-the-art prediction performances over five properties out of six with up to 10\% improvement. Our work shows that to deal with the high complexity of mapping the crystal materials structures to their properties, large-scale very deep graph neural networks are needed to achieve robust performances.