 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdversarial Examples Might be Avoidable: The Role of Data Concentration in Adversarial Robustness
Sep 28, 2023



The susceptibility of modern machine learning classifiers to adversarial examples has motivated theoretical results suggesting that these might be unavoidable. However, these results can be too general to be applicable to natural data distributions. Indeed, humans are quite robust for tasks involving vision. This apparent conflict motivates a deeper dive into the question: Are adversarial examples truly unavoidable? In this work, we theoretically demonstrate that a key property of the data distribution -- concentration on small-volume subsets of the input space -- determines whether a robust classifier exists. We further demonstrate that, for a data distribution concentrated on a union of low-dimensional linear subspaces, exploiting data structure naturally leads to classifiers that enjoy good robustness guarantees, improving upon methods for provable certification in certain regimes.
Sparsity-aware generalization theory for deep neural networks
Jul 04, 2023Deep artificial neural networks achieve surprising generalization abilities that remain poorly understood. In this paper, we present a new approach to analyzing generalization for deep feed-forward ReLU networks that takes advantage of the degree of sparsity that is achieved in the hidden layer activations. By developing a framework that accounts for this reduced effective model size for each input sample, we are able to show fundamental trade-offs between sparsity and generalization. Importantly, our results make no strong assumptions about the degree of sparsity achieved by the model, and it improves over recent norm-based approaches. We illustrate our results numerically, demonstrating non-vacuous bounds when coupled with data-dependent priors in specific settings, even in over-parametrized models.
Understanding Noise-Augmented Training for Randomized Smoothing
May 08, 2023



Randomized smoothing is a technique for providing provable robustness guarantees against adversarial attacks while making minimal assumptions about a classifier. This method relies on taking a majority vote of any base classifier over multiple noise-perturbed inputs to obtain a smoothed classifier, and it remains the tool of choice to certify deep and complex neural network models. Nonetheless, non-trivial performance of such smoothed classifier crucially depends on the base model being trained on noise-augmented data, i.e., on a smoothed input distribution. While widely adopted in practice, it is still unclear how this noisy training of the base classifier precisely affects the risk of the robust smoothed classifier, leading to heuristics and tricks that are poorly understood. In this work we analyze these trade-offs theoretically in a binary classification setting, proving that these common observations are not universal. We show that, without making stronger distributional assumptions, no benefit can be expected from predictors trained with noise-augmentation, and we further characterize distributions where such benefit is obtained. Our analysis has direct implications to the practical deployment of randomized smoothing, and we illustrate some of these via experiments on CIFAR-10 and MNIST, as well as on synthetic datasets.
How to Trust Your Diffusion Model: A Convex Optimization Approach to Conformal Risk Control
Feb 07, 2023Score-based generative modeling, informally referred to as diffusion models, continue to grow in popularity across several important domains and tasks. While they provide high-quality and diverse samples from empirical distributions, important questions remain on the reliability and trustworthiness of these sampling procedures for their responsible use in critical scenarios. Conformal prediction is a modern tool to construct finite-sample, distribution-free uncertainty guarantees for any black-box predictor. In this work, we focus on image-to-image regression tasks and we present a generalization of the Risk-Controlling Prediction Sets (RCPS) procedure, that we term $K$-RCPS, which allows to $(i)$ provide entrywise calibrated intervals for future samples of any diffusion model, and $(ii)$ control a certain notion of risk with respect to a ground truth image with minimal mean interval length. Differently from existing conformal risk control procedures, ours relies on a novel convex optimization approach that allows for multidimensional risk control while provably minimizing the mean interval length. We illustrate our approach on two real-world image denoising problems: on natural images of faces as well as on computed tomography (CT) scans of the abdomen, demonstrating state of the art performance.
Weakly Supervised Learning Significantly Reduces the Number of Labels Required for Intracranial Hemorrhage Detection on Head CT
Nov 29, 2022



Modern machine learning pipelines, in particular those based on deep learning (DL) models, require large amounts of labeled data. For classification problems, the most common learning paradigm consists of presenting labeled examples during training, thus providing strong supervision on what constitutes positive and negative samples. This constitutes a major obstacle for the development of DL models in radiology--in particular for cross-sectional imaging (e.g., computed tomography [CT] scans)--where labels must come from manual annotations by expert radiologists at the image or slice-level. These differ from examination-level annotations, which are coarser but cheaper, and could be extracted from radiology reports using natural language processing techniques. This work studies the question of what kind of labels should be collected for the problem of intracranial hemorrhage detection in brain CT. We investigate whether image-level annotations should be preferred to examination-level ones. By framing this task as a multiple instance learning problem, and employing modern attention-based DL architectures, we analyze the degree to which different levels of supervision improve detection performance. We find that strong supervision (i.e., learning with local image-level annotations) and weak supervision (i.e., learning with only global examination-level labels) achieve comparable performance in examination-level hemorrhage detection (the task of selecting the images in an examination that show signs of hemorrhage) as well as in image-level hemorrhage detection (highlighting those signs within the selected images). Furthermore, we study this behavior as a function of the number of labels available during training. Our results suggest that local labels may not be necessary at all for these tasks, drastically reducing the time and cost involved in collecting and curating datasets.
DeepSTI: Towards Tensor Reconstruction using Fewer Orientations in Susceptibility Tensor Imaging
Sep 09, 2022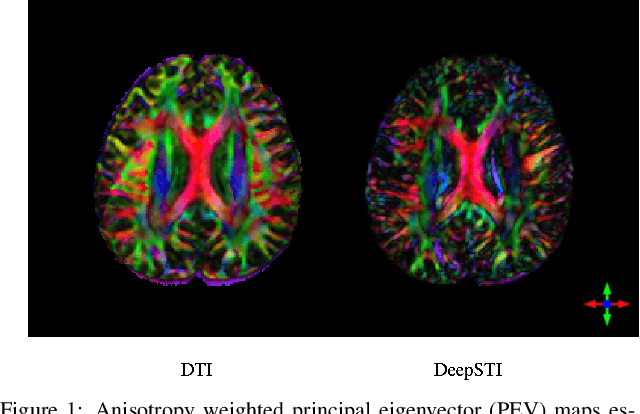
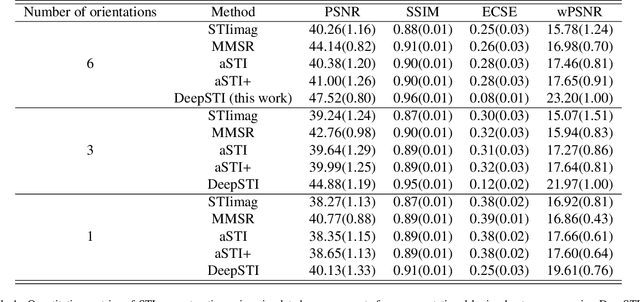
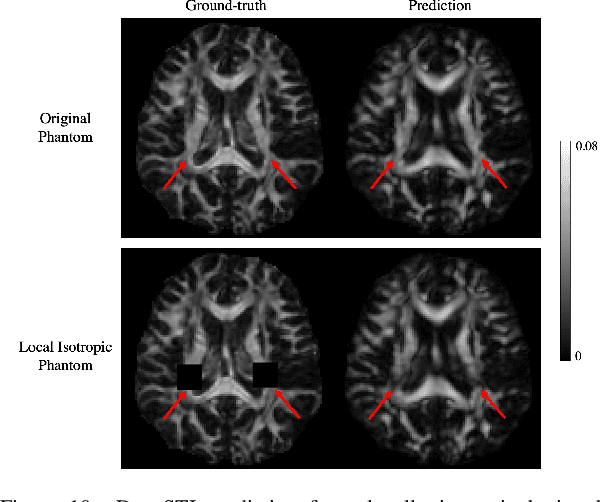
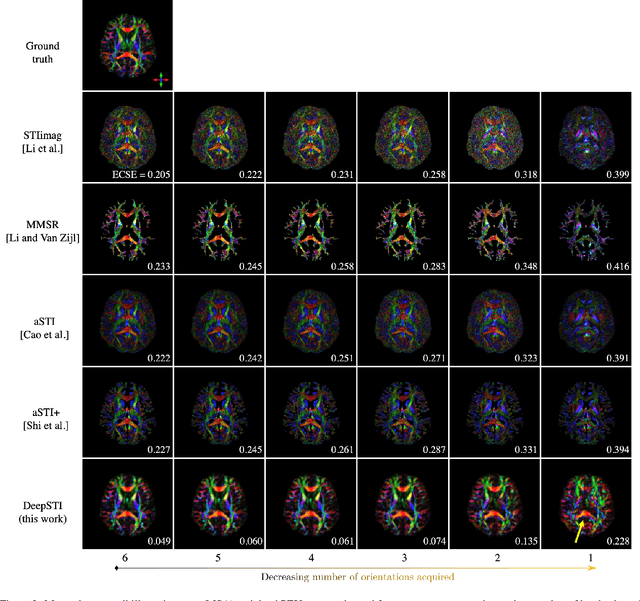
Susceptibility tensor imaging (STI) is an emerging magnetic resonance imaging technique that characterizes the anisotropic tissue magnetic susceptibility with a second-order tensor model. STI has the potential to provide information for both the reconstruction of white matter fiber pathways and detection of myelin changes in the brain at mm resolution or less, which would be of great value for understanding brain structure and function in healthy and diseased brain. However, the application of STI in vivo has been hindered by its cumbersome and time-consuming acquisition requirement of measuring susceptibility induced MR phase changes at multiple (usually more than six) head orientations. This complexity is enhanced by the limitation in head rotation angles due to physical constraints of the head coil. As a result, STI has not yet been widely applied in human studies in vivo. In this work, we tackle these issues by proposing an image reconstruction algorithm for STI that leverages data-driven priors. Our method, called DeepSTI, learns the data prior implicitly via a deep neural network that approximates the proximal operator of a regularizer function for STI. The dipole inversion problem is then solved iteratively using the learned proximal network. Experimental results using both simulation and in vivo human data demonstrate great improvement over state-of-the-art algorithms in terms of the reconstructed tensor image, principal eigenvector maps and tractography results, while allowing for tensor reconstruction with MR phase measured at much less than six different orientations. Notably, promising reconstruction results are achieved by our method from only one orientation in human in vivo, and we demonstrate a potential application of this technique for estimating lesion susceptibility anisotropy in patients with multiple sclerosis.
Estimating and Controlling for Fairness via Sensitive Attribute Predictors
Jul 25, 2022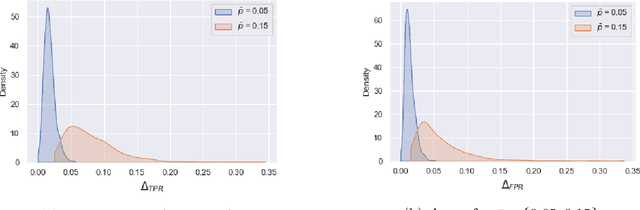
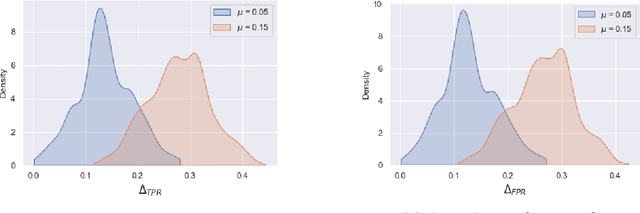
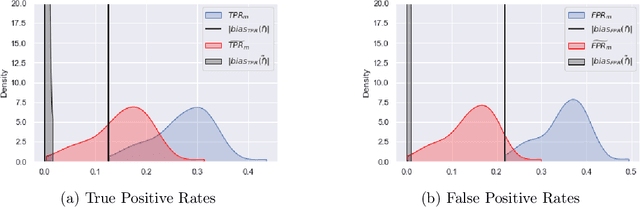
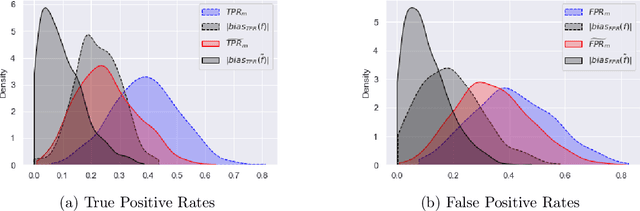
Although machine learning classifiers have been increasingly used in high-stakes decision making (e.g., cancer diagnosis, criminal prosecution decisions), they have demonstrated biases against underrepresented groups. Standard definitions of fairness require access to sensitive attributes of interest (e.g., gender and race), which are often unavailable. In this work we demonstrate that in these settings where sensitive attributes are unknown, one can still reliably estimate and ultimately control for fairness by using proxy sensitive attributes derived from a sensitive attribute predictor. Specifically, we first show that with just a little knowledge of the complete data distribution, one may use a sensitive attribute predictor to obtain upper and lower bounds of the classifier's true fairness metric. Second, we demonstrate how one can provably control for fairness with respect to the true sensitive attributes by controlling for fairness with respect to the proxy sensitive attributes. Our results hold under assumptions that are significantly milder than previous works. We illustrate our results on a series of synthetic and real datasets.
From Shapley back to Pearson: Hypothesis Testing via the Shapley Value
Jul 14, 2022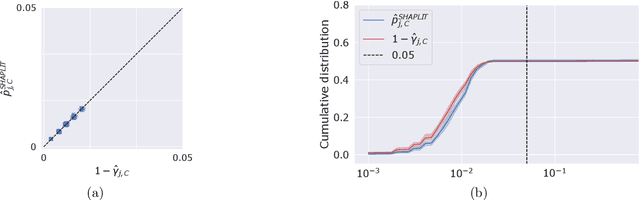
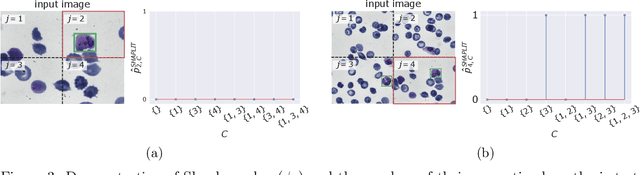
Machine learning models, in particular artificial neural networks, are increasingly used to inform decision making in high-stakes scenarios across a variety of fields--from financial services, to public safety, and healthcare. While neural networks have achieved remarkable performance in many settings, their complex nature raises concerns on their reliability, trustworthiness, and fairness in real-world scenarios. As a result, several a-posteriori explanation methods have been proposed to highlight the features that influence a model's prediction. Notably, the Shapley value--a game theoretic quantity that satisfies several desirable properties--has gained popularity in the machine learning explainability literature. More traditionally, however, feature importance in statistical learning has been formalized by conditional independence, and a standard way to test for it is via Conditional Randomization Tests (CRTs). So far, these two perspectives on interpretability and feature importance have been considered distinct and separate. In this work, we show that Shapley-based explanation methods and conditional independence testing for feature importance are closely related. More precisely, we prove that evaluating a Shapley coefficient amounts to performing a specific set of conditional independence tests, as implemented by a procedure similar to the CRT but for a different null hypothesis. Furthermore, the obtained game-theoretic values upper bound the $p$-values of such tests. As a result, we grant large Shapley coefficients with a precise statistical sense of importance with controlled type I error.
Adversarial robustness of sparse local Lipschitz predictors
Feb 26, 2022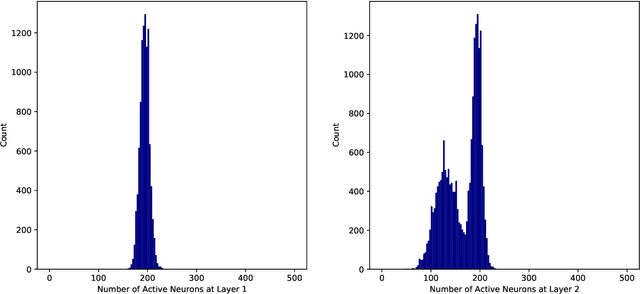
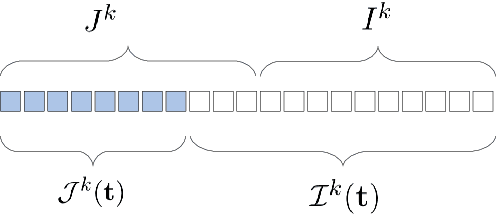
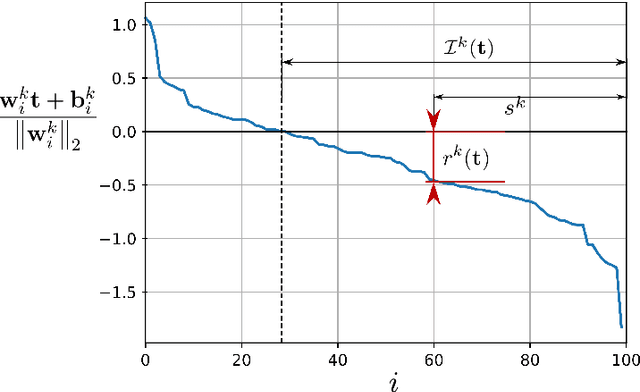
This work studies the adversarial robustness of parametric functions composed of a linear predictor and a non-linear representation map. Our analysis relies on sparse local Lipschitzness (SLL), an extension of local Lipschitz continuity that better captures the stability and reduced effective dimensionality of predictors upon local perturbations. SLL functions preserve a certain degree of structure, given by the sparsity pattern in the representation map, and include several popular hypothesis classes, such as piece-wise linear models, Lasso and its variants, and deep feed-forward ReLU networks. We provide a tighter robustness certificate on the minimal energy of an adversarial example, as well as tighter data-dependent non-uniform bounds on the robust generalization error of these predictors. We instantiate these results for the case of deep neural networks and provide numerical evidence that supports our results, shedding new insights into natural regularization strategies to increase the robustness of these models.
Entrywise Recovery Guarantees for Sparse PCA via Sparsistent Algorithms
Feb 08, 2022Sparse Principal Component Analysis (PCA) is a prevalent tool across a plethora of subfields of applied statistics. While several results have characterized the recovery error of the principal eigenvectors, these are typically in spectral or Frobenius norms. In this paper, we provide entrywise $\ell_{2,\infty}$ bounds for Sparse PCA under a general high-dimensional subgaussian design. In particular, our results hold for any algorithm that selects the correct support with high probability, those that are sparsistent. Our bound improves upon known results by providing a finer characterization of the estimation error, and our proof uses techniques recently developed for entrywise subspace perturbation theory.