 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph AI generates neurological hypotheses validated in molecular, organoid, and clinical systems
Dec 13, 2025Neurological diseases are the leading global cause of disability, yet most lack disease-modifying treatments. We present PROTON, a heterogeneous graph transformer that generates testable hypotheses across molecular, organoid, and clinical systems. To evaluate PROTON, we apply it to Parkinson's disease (PD), bipolar disorder (BD), and Alzheimer's disease (AD). In PD, PROTON linked genetic risk loci to genes essential for dopaminergic neuron survival and predicted pesticides toxic to patient-derived neurons, including the insecticide endosulfan, which ranked within the top 1.29% of predictions. In silico screens performed by PROTON reproduced six genome-wide $α$-synuclein experiments, including a split-ubiquitin yeast two-hybrid system (normalized enrichment score [NES] = 2.30, FDR-adjusted $p < 1 \times 10^{-4}$), an ascorbate peroxidase proximity labeling assay (NES = 2.16, FDR $< 1 \times 10^{-4}$), and a high-depth targeted exome sequencing study in 496 synucleinopathy patients (NES = 2.13, FDR $< 1 \times 10^{-4}$). In BD, PROTON predicted calcitriol as a candidate drug that reversed proteomic alterations observed in cortical organoids derived from BD patients. In AD, we evaluated PROTON predictions in health records from $n = 610,524$ patients at Mass General Brigham, confirming that five PROTON-predicted drugs were associated with reduced seven-year dementia risk (minimum hazard ratio = 0.63, 95% CI: 0.53-0.75, $p < 1 \times 10^{-7}$). PROTON generated neurological hypotheses that were evaluated across molecular, organoid, and clinical systems, defining a path for AI-driven discovery in neurological disease.
Variational auto-encoding of protein sequences
Jan 03, 2018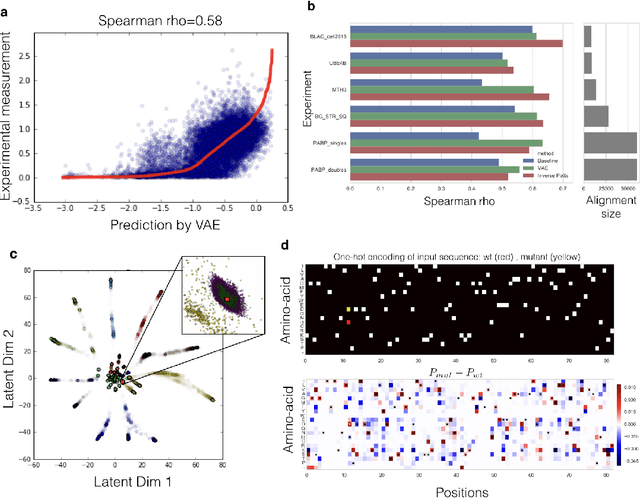
Proteins are responsible for the most diverse set of functions in biology. The ability to extract information from protein sequences and to predict the effects of mutations is extremely valuable in many domains of biology and medicine. However the mapping between protein sequence and function is complex and poorly understood. Here we present an embedding of natural protein sequences using a Variational Auto-Encoder and use it to predict how mutations affect protein function. We use this unsupervised approach to cluster natural variants and learn interactions between sets of positions within a protein. This approach generally performs better than baseline methods that consider no interactions within sequences, and in some cases better than the state-of-the-art approaches that use the inverse-Potts model. This generative model can be used to computationally guide exploration of protein sequence space and to better inform rational and automatic protein design.
Puzzle Imaging: Using Large-scale Dimensionality Reduction Algorithms for Localization
Jun 21, 2015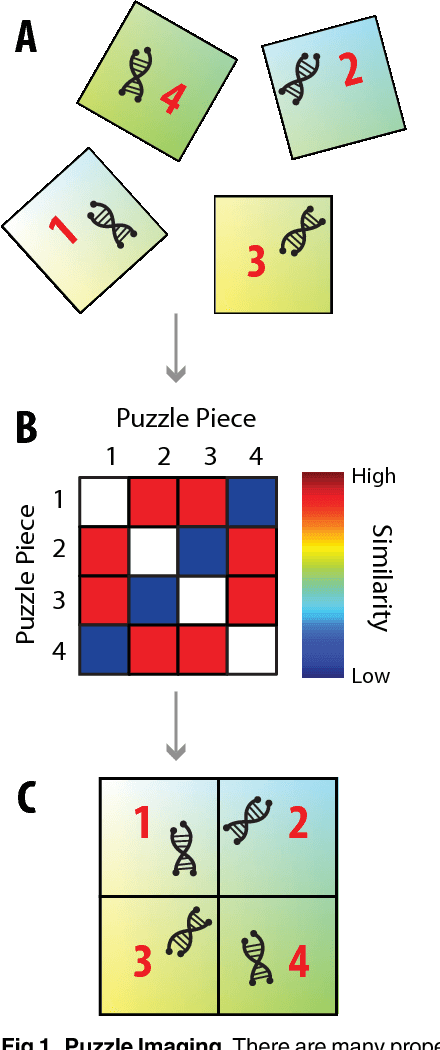

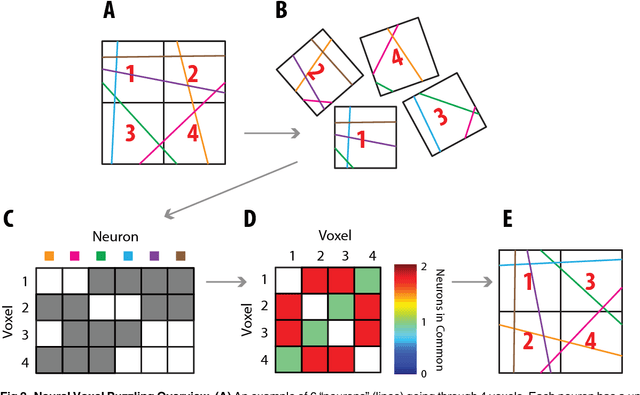
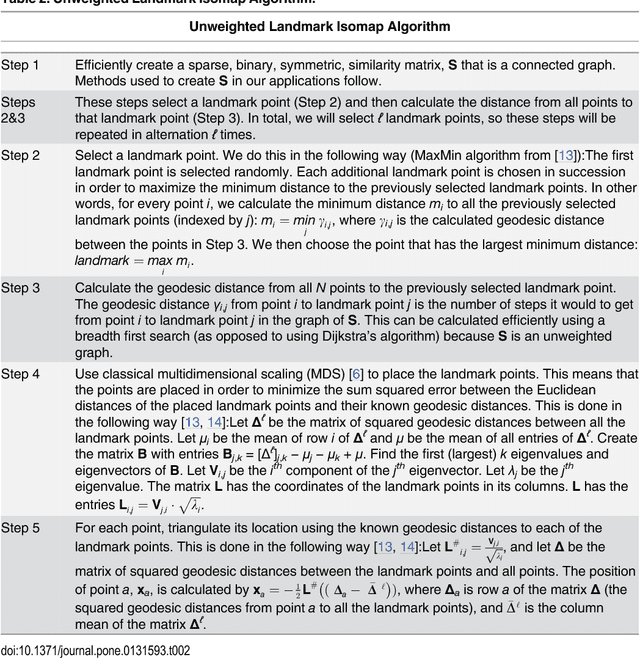
Current high-resolution imaging techniques require an intact sample that preserves spatial relationships. We here present a novel approach, "puzzle imaging," that allows imaging a spatially scrambled sample. This technique takes many spatially disordered samples, and then pieces them back together using local properties embedded within the sample. We show that puzzle imaging can efficiently produce high-resolution images using dimensionality reduction algorithms. We demonstrate the theoretical capabilities of puzzle imaging in three biological scenarios, showing that (1) relatively precise 3-dimensional brain imaging is possible; (2) the physical structure of a neural network can often be recovered based only on the neural connectivity matrix; and (3) a chemical map could be reproduced using bacteria with chemosensitive DNA and conjugative transfer. The ability to reconstruct scrambled images promises to enable imaging based on DNA sequencing of homogenized tissue samples.