 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-Improvement Imitation with Biologically Guided Search for Protein Design Under Oracle Budgets
May 26, 2026Protein sequence optimization under tight oracle budgets requires methods that explore vast combinatorial spaces while making each evaluation informative. Existing reinforcement learning and off-policy generative approaches often degrade under surrogate noise, and position-agnostic mutation proposals risk disrupting functionally critical residues. We introduce SILO, a trajectory-level self-improvement imitation framework for oracle-budgeted protein design. SILO uses a hierarchical edit policy that decomposes each mutation into a position choice followed by a residue choice. In each active-learning round, the policy samples candidate trajectories via incremental stochastic beam search without replacement (SBS), and a UCB-based proxy ensemble, combined with an alanine-scan fitness score (AFS), selects candidates with functionally relevant edits for in silico oracle evaluation. The policy is then updated by next-action cross-entropy imitation on the round's best oracle-labeled trajectories, avoiding value-function estimation. Across eight reproduced protein fitness landscapes and five strong baselines from prior work, SILO achieves the highest maximum and top-100 mean fitness on 8 of 8 landscapes within our evaluations, often exhibiting faster early-stage improvement. In low-data and noisy-proxy stress tests on two landscapes per setting, SILO remains competitive or best when several baselines degrade. Ablations show that SBS with AFS account for much of the gains, with iterative imitation providing additional improvement. Code is available at: https://github.com/grimmlab/SILO.git
Efficient network-guided multi-locus association mapping with graph cuts
Apr 18, 2013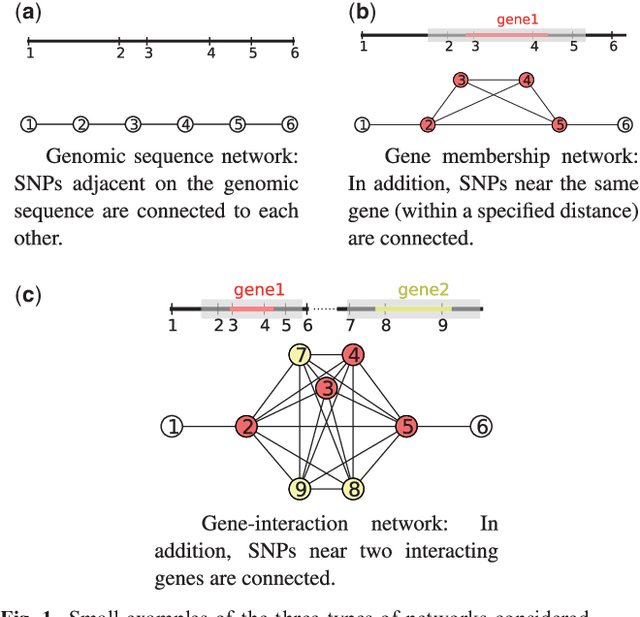
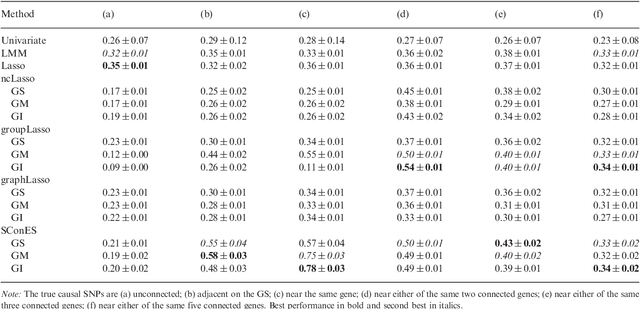
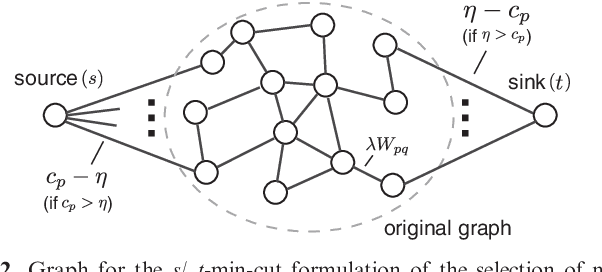
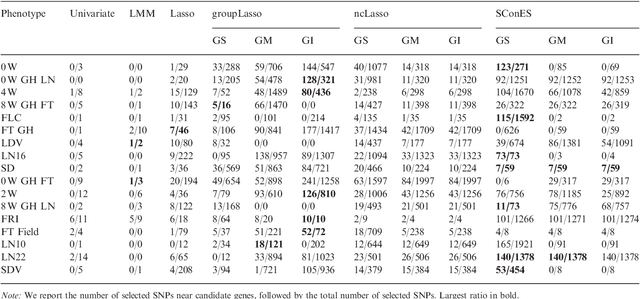
As an increasing number of genome-wide association studies reveal the limitations of attempting to explain phenotypic heritability by single genetic loci, there is growing interest for associating complex phenotypes with sets of genetic loci. While several methods for multi-locus mapping have been proposed, it is often unclear how to relate the detected loci to the growing knowledge about gene pathways and networks. The few methods that take biological pathways or networks into account are either restricted to investigating a limited number of predetermined sets of loci, or do not scale to genome-wide settings. We present SConES, a new efficient method to discover sets of genetic loci that are maximally associated with a phenotype, while being connected in an underlying network. Our approach is based on a minimum cut reformulation of the problem of selecting features under sparsity and connectivity constraints that can be solved exactly and rapidly. SConES outperforms state-of-the-art competitors in terms of runtime, scales to hundreds of thousands of genetic loci, and exhibits higher power in detecting causal SNPs in simulation studies than existing methods. On flowering time phenotypes and genotypes from Arabidopsis thaliana, SConES detects loci that enable accurate phenotype prediction and that are supported by the literature. Matlab code for SConES is available at http://webdav.tuebingen.mpg.de/u/karsten/Forschung/scones/
Geometric tree kernels: Classification of COPD from airway tree geometry
Apr 08, 2013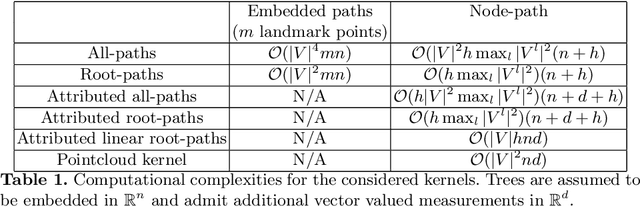

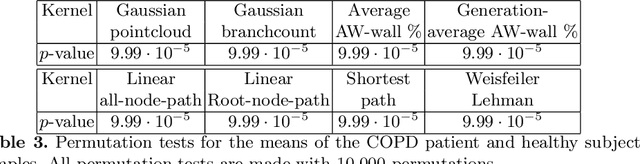
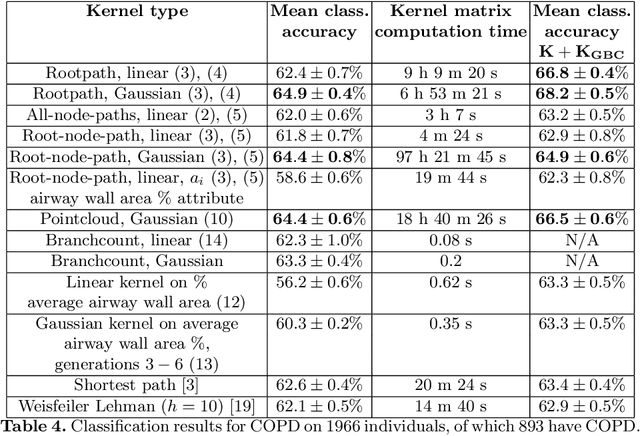
Methodological contributions: This paper introduces a family of kernels for analyzing (anatomical) trees endowed with vector valued measurements made along the tree. While state-of-the-art graph and tree kernels use combinatorial tree/graph structure with discrete node and edge labels, the kernels presented in this paper can include geometric information such as branch shape, branch radius or other vector valued properties. In addition to being flexible in their ability to model different types of attributes, the presented kernels are computationally efficient and some of them can easily be computed for large datasets (N of the order 10.000) of trees with 30-600 branches. Combining the kernels with standard machine learning tools enables us to analyze the relation between disease and anatomical tree structure and geometry. Experimental results: The kernels are used to compare airway trees segmented from low-dose CT, endowed with branch shape descriptors and airway wall area percentage measurements made along the tree. Using kernelized hypothesis testing we show that the geometric airway trees are significantly differently distributed in patients with Chronic Obstructive Pulmonary Disease (COPD) than in healthy individuals. The geometric tree kernels also give a significant increase in the classification accuracy of COPD from geometric tree structure endowed with airway wall thickness measurements in comparison with state-of-the-art methods, giving further insight into the relationship between airway wall thickness and COPD. Software: Software for computing kernels and statistical tests is available at http://image.diku.dk/aasa/software.php.