 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMassSpecGym: A benchmark for the discovery and identification of molecules
Oct 30, 2024The discovery and identification of molecules in biological and environmental samples is crucial for advancing biomedical and chemical sciences. Tandem mass spectrometry (MS/MS) is the leading technique for high-throughput elucidation of molecular structures. However, decoding a molecular structure from its mass spectrum is exceptionally challenging, even when performed by human experts. As a result, the vast majority of acquired MS/MS spectra remain uninterpreted, thereby limiting our understanding of the underlying (bio)chemical processes. Despite decades of progress in machine learning applications for predicting molecular structures from MS/MS spectra, the development of new methods is severely hindered by the lack of standard datasets and evaluation protocols. To address this problem, we propose MassSpecGym -- the first comprehensive benchmark for the discovery and identification of molecules from MS/MS data. Our benchmark comprises the largest publicly available collection of high-quality labeled MS/MS spectra and defines three MS/MS annotation challenges: \textit{de novo} molecular structure generation, molecule retrieval, and spectrum simulation. It includes new evaluation metrics and a generalization-demanding data split, therefore standardizing the MS/MS annotation tasks and rendering the problem accessible to the broad machine learning community. MassSpecGym is publicly available at \url{https://github.com/pluskal-lab/MassSpecGym}.
Accurate, fully-automated NMR spectral profiling for metabolomics
Sep 08, 2014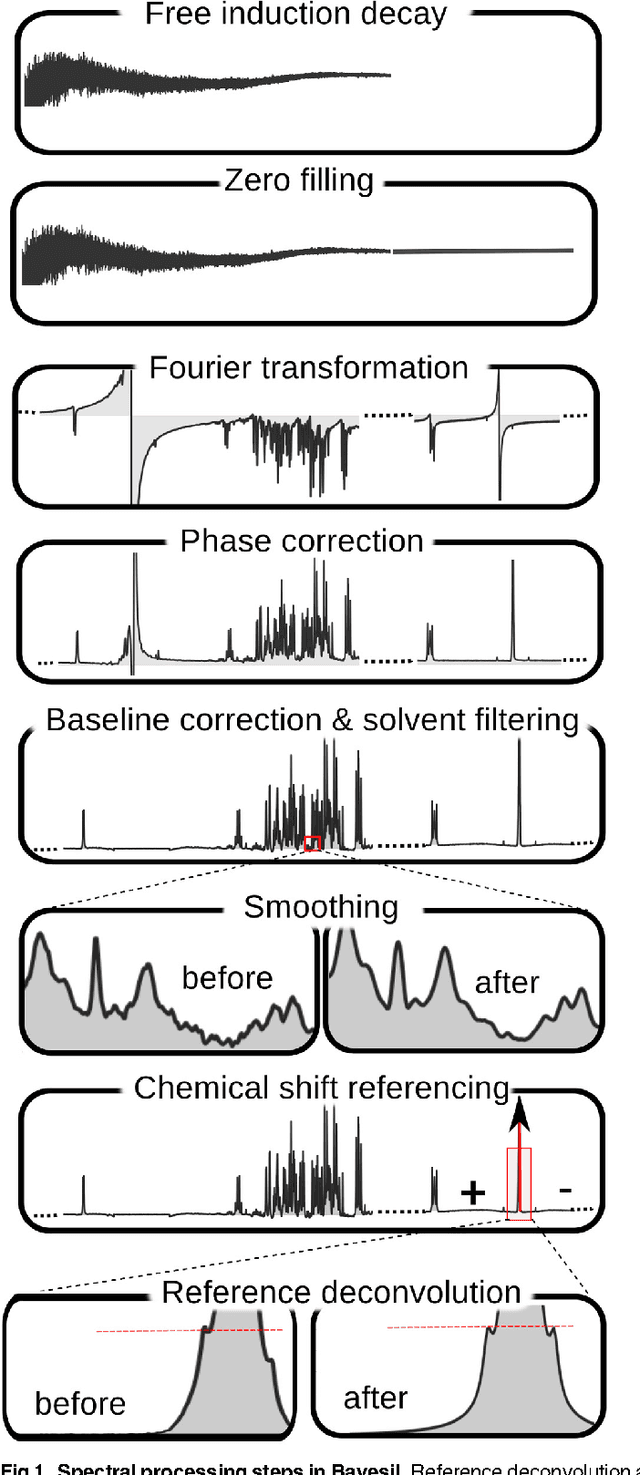
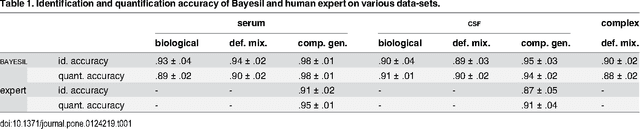
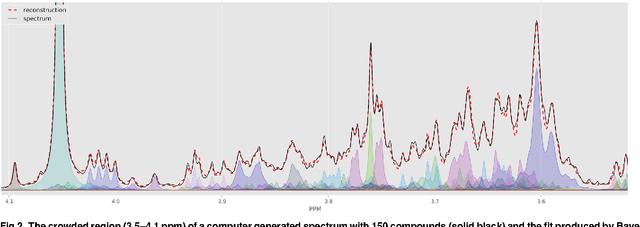
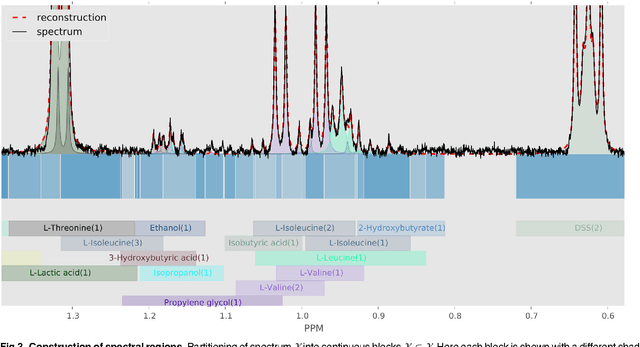
Many diseases cause significant changes to the concentrations of small molecules (aka metabolites) that appear in a person's biofluids, which means such diseases can often be readily detected from a person's "metabolic profile". This information can be extracted from a biofluid's NMR spectrum. Today, this is often done manually by trained human experts, which means this process is relatively slow, expensive and error-prone. This paper presents a tool, Bayesil, that can quickly, accurately and autonomously produce a complex biofluid's (e.g., serum or CSF) metabolic profile from a 1D1H NMR spectrum. This requires first performing several spectral processing steps then matching the resulting spectrum against a reference compound library, which contains the "signatures" of each relevant metabolite. Many of these steps are novel algorithms and our matching step views spectral matching as an inference problem within a probabilistic graphical model that rapidly approximates the most probable metabolic profile. Our extensive studies on a diverse set of complex mixtures, show that Bayesil can autonomously find the concentration of all NMR-detectable metabolites accurately (~90% correct identification and ~10% quantification error), in <5minutes on a single CPU. These results demonstrate that Bayesil is the first fully-automatic publicly-accessible system that provides quantitative NMR spectral profiling effectively -- with an accuracy that meets or exceeds the performance of trained experts. We anticipate this tool will usher in high-throughput metabolomics and enable a wealth of new applications of NMR in clinical settings. Available at http://www.bayesil.ca.