 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSticking the Landing: Simple, Lower-Variance Gradient Estimators for Variational Inference
May 28, 2017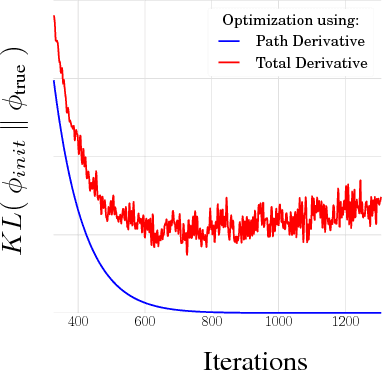
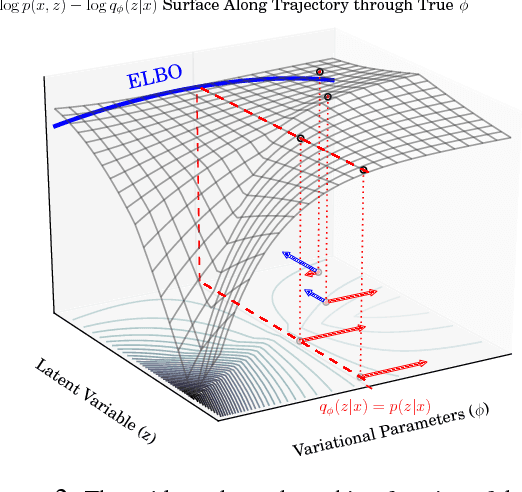
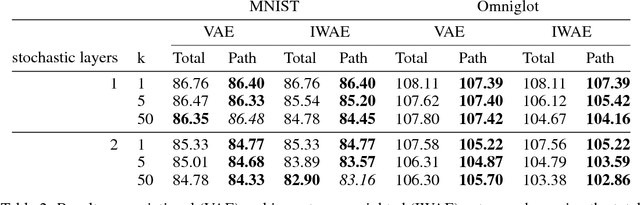
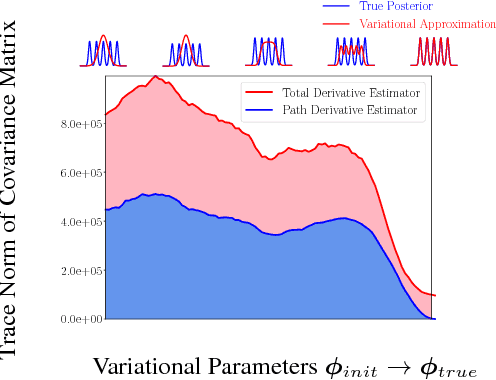
We propose a simple and general variant of the standard reparameterized gradient estimator for the variational evidence lower bound. Specifically, we remove a part of the total derivative with respect to the variational parameters that corresponds to the score function. Removing this term produces an unbiased gradient estimator whose variance approaches zero as the approximate posterior approaches the exact posterior. We analyze the behavior of this gradient estimator theoretically and empirically, and generalize it to more complex variational distributions such as mixtures and importance-weighted posteriors.
Neural networks for the prediction organic chemistry reactions
Oct 17, 2016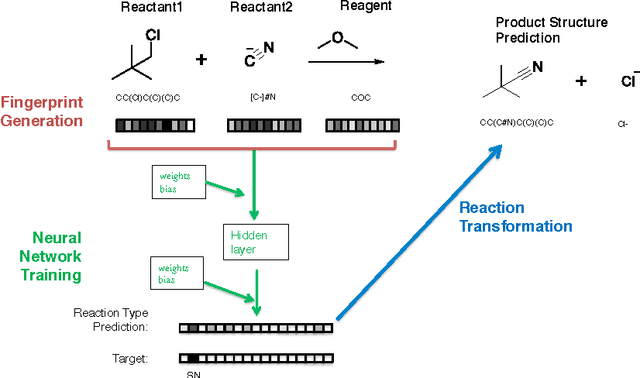
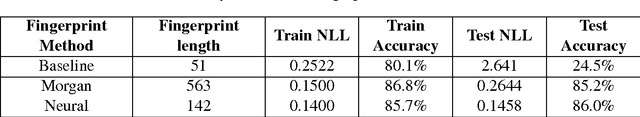
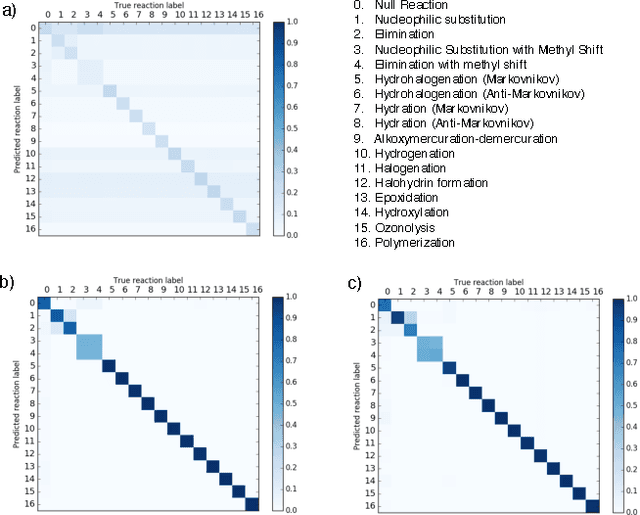
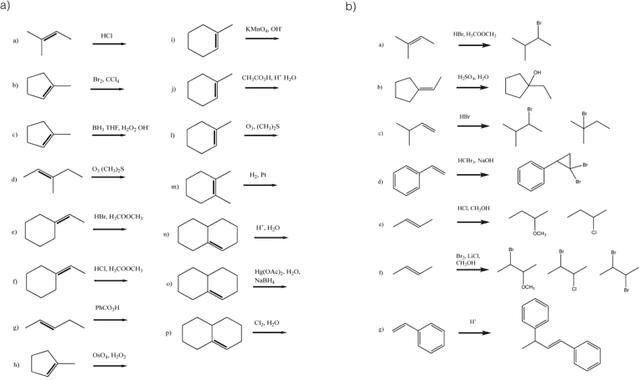
Reaction prediction remains one of the major challenges for organic chemistry, and is a pre-requisite for efficient synthetic planning. It is desirable to develop algorithms that, like humans, "learn" from being exposed to examples of the application of the rules of organic chemistry. We explore the use of neural networks for predicting reaction types, using a new reaction fingerprinting method. We combine this predictor with SMARTS transformations to build a system which, given a set of reagents and re- actants, predicts the likely products. We test this method on problems from a popular organic chemistry textbook.
* 21 pages, 5 figures
Optimally-Weighted Herding is Bayesian Quadrature
Jul 15, 2016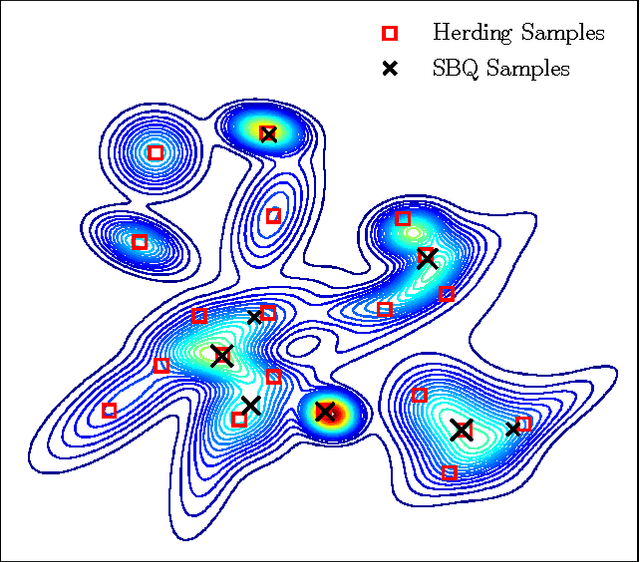
Herding and kernel herding are deterministic methods of choosing samples which summarise a probability distribution. A related task is choosing samples for estimating integrals using Bayesian quadrature. We show that the criterion minimised when selecting samples in kernel herding is equivalent to the posterior variance in Bayesian quadrature. We then show that sequential Bayesian quadrature can be viewed as a weighted version of kernel herding which achieves performance superior to any other weighted herding method. We demonstrate empirically a rate of convergence faster than O(1/N). Our results also imply an upper bound on the empirical error of the Bayesian quadrature estimate.
Avoiding pathologies in very deep networks
Jul 08, 2016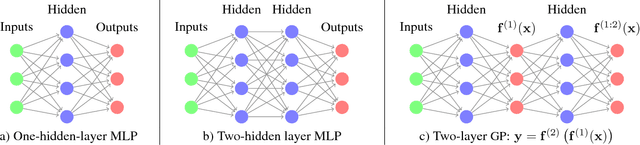
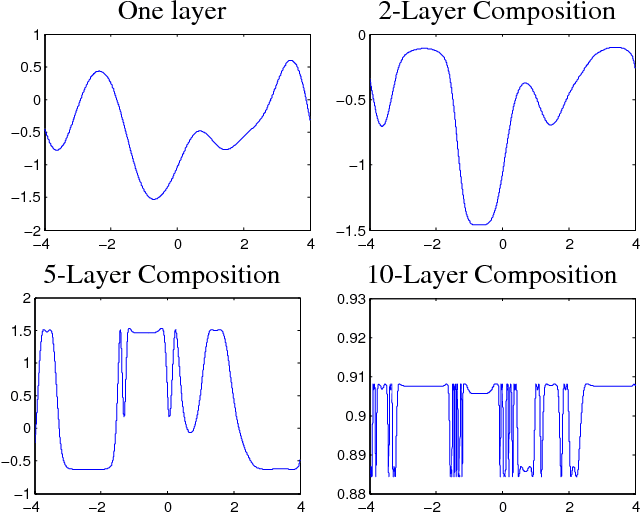
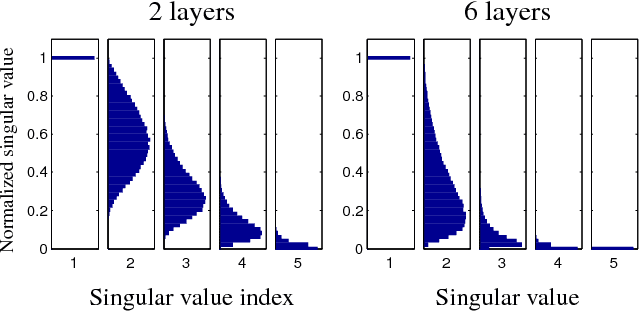
Choosing appropriate architectures and regularization strategies for deep networks is crucial to good predictive performance. To shed light on this problem, we analyze the analogous problem of constructing useful priors on compositions of functions. Specifically, we study the deep Gaussian process, a type of infinitely-wide, deep neural network. We show that in standard architectures, the representational capacity of the network tends to capture fewer degrees of freedom as the number of layers increases, retaining only a single degree of freedom in the limit. We propose an alternate network architecture which does not suffer from this pathology. We also examine deep covariance functions, obtained by composing infinitely many feature transforms. Lastly, we characterize the class of models obtained by performing dropout on Gaussian processes.
Convolutional Networks on Graphs for Learning Molecular Fingerprints
Nov 03, 2015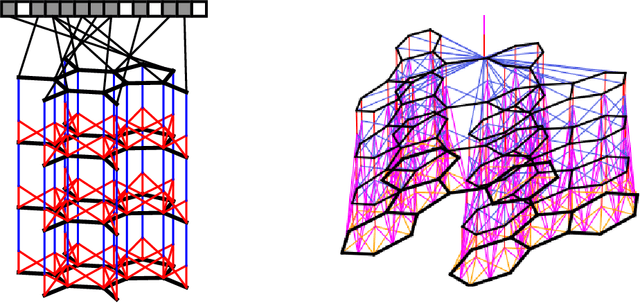
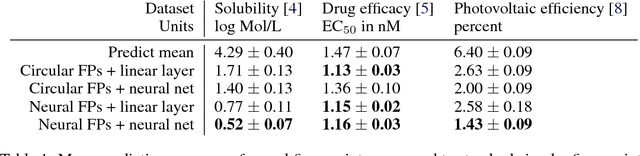

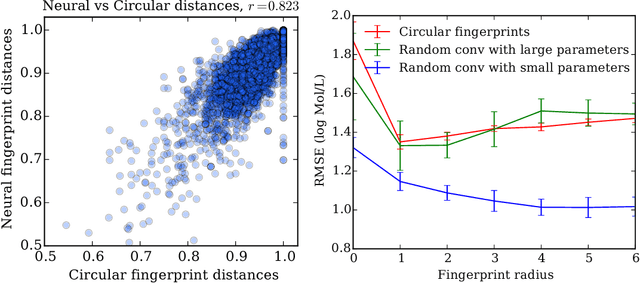
We introduce a convolutional neural network that operates directly on graphs. These networks allow end-to-end learning of prediction pipelines whose inputs are graphs of arbitrary size and shape. The architecture we present generalizes standard molecular feature extraction methods based on circular fingerprints. We show that these data-driven features are more interpretable, and have better predictive performance on a variety of tasks.
Early Stopping is Nonparametric Variational Inference
Apr 06, 2015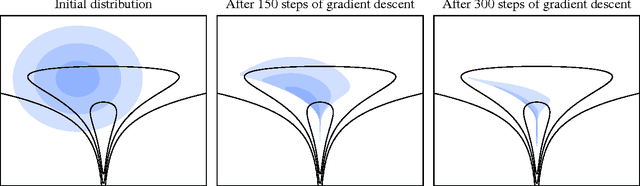
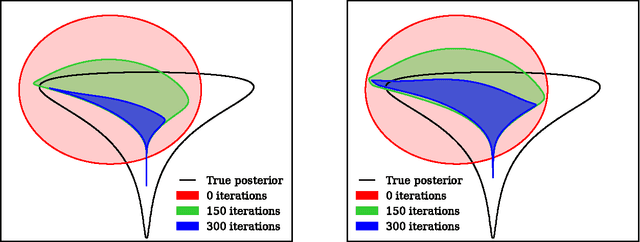
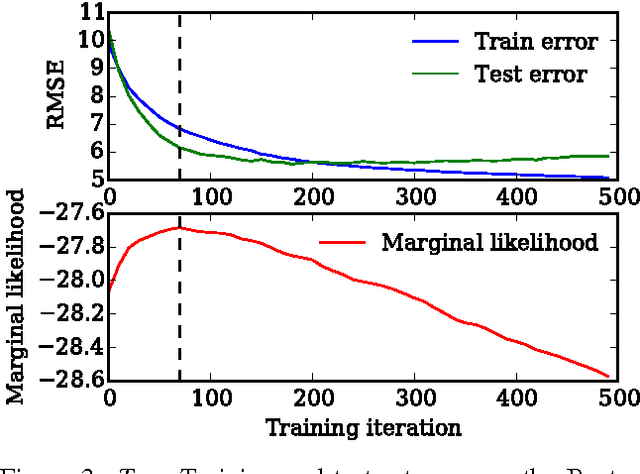
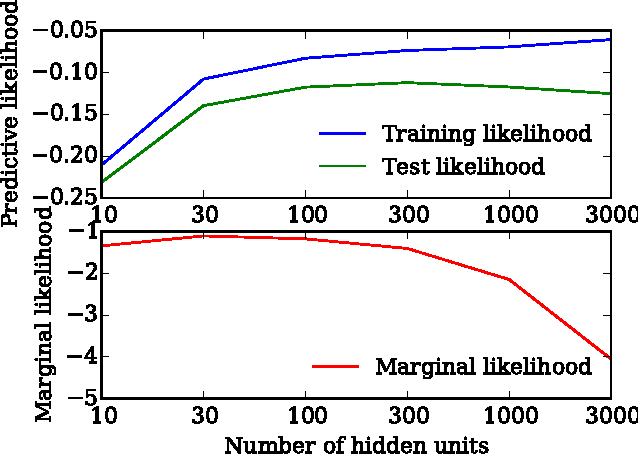
We show that unconverged stochastic gradient descent can be interpreted as a procedure that samples from a nonparametric variational approximate posterior distribution. This distribution is implicitly defined as the transformation of an initial distribution by a sequence of optimization updates. By tracking the change in entropy over this sequence of transformations during optimization, we form a scalable, unbiased estimate of the variational lower bound on the log marginal likelihood. We can use this bound to optimize hyperparameters instead of using cross-validation. This Bayesian interpretation of SGD suggests improved, overfitting-resistant optimization procedures, and gives a theoretical foundation for popular tricks such as early stopping and ensembling. We investigate the properties of this marginal likelihood estimator on neural network models.
Gradient-based Hyperparameter Optimization through Reversible Learning
Apr 02, 2015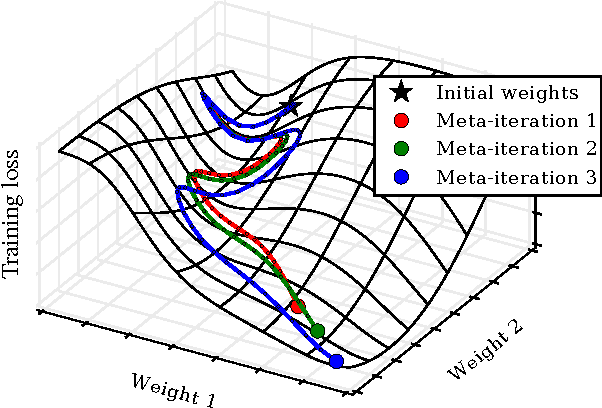
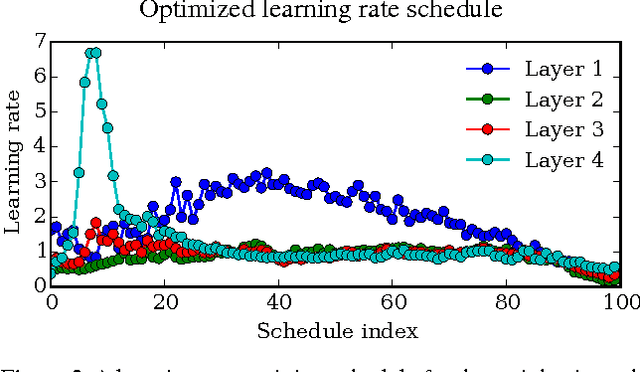
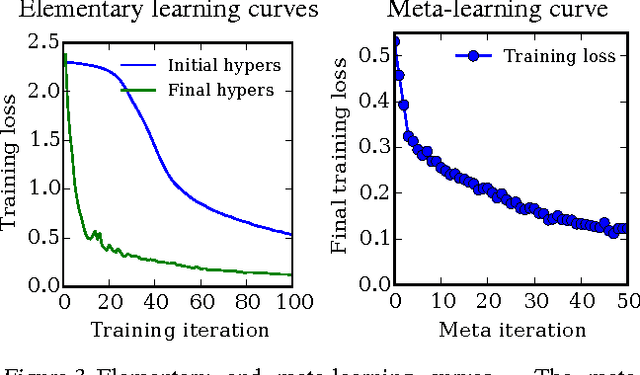
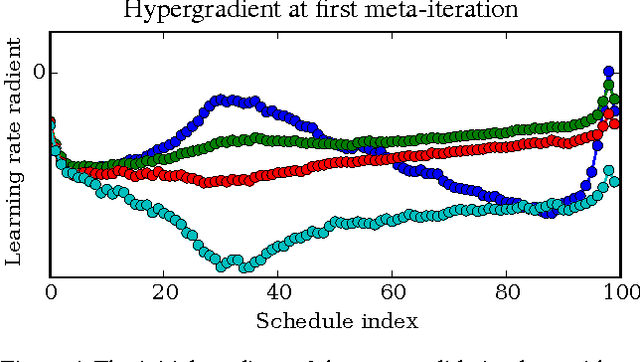
Tuning hyperparameters of learning algorithms is hard because gradients are usually unavailable. We compute exact gradients of cross-validation performance with respect to all hyperparameters by chaining derivatives backwards through the entire training procedure. These gradients allow us to optimize thousands of hyperparameters, including step-size and momentum schedules, weight initialization distributions, richly parameterized regularization schemes, and neural network architectures. We compute hyperparameter gradients by exactly reversing the dynamics of stochastic gradient descent with momentum.
Probabilistic ODE Solvers with Runge-Kutta Means
Oct 24, 2014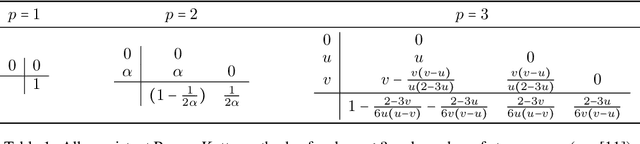
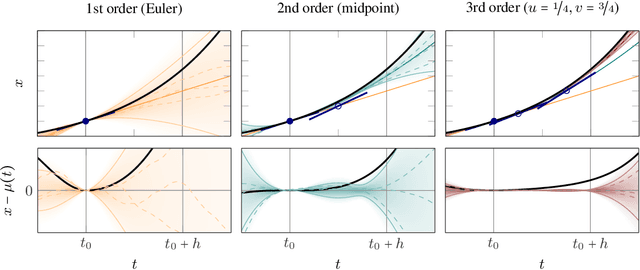
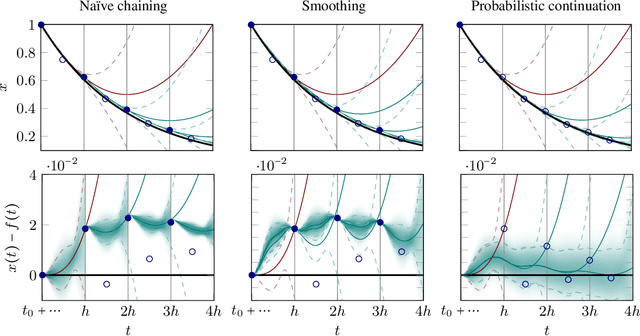
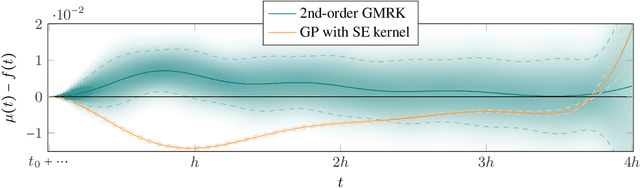
Runge-Kutta methods are the classic family of solvers for ordinary differential equations (ODEs), and the basis for the state of the art. Like most numerical methods, they return point estimates. We construct a family of probabilistic numerical methods that instead return a Gauss-Markov process defining a probability distribution over the ODE solution. In contrast to prior work, we construct this family such that posterior means match the outputs of the Runge-Kutta family exactly, thus inheriting their proven good properties. Remaining degrees of freedom not identified by the match to Runge-Kutta are chosen such that the posterior probability measure fits the observed structure of the ODE. Our results shed light on the structure of Runge-Kutta solvers from a new direction, provide a richer, probabilistic output, have low computational cost, and raise new research questions.
Raiders of the Lost Architecture: Kernels for Bayesian Optimization in Conditional Parameter Spaces
Sep 14, 2014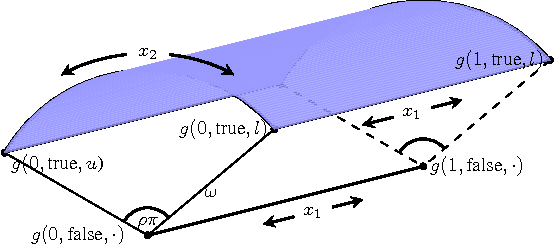

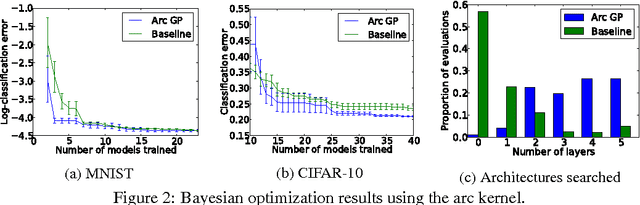
In practical Bayesian optimization, we must often search over structures with differing numbers of parameters. For instance, we may wish to search over neural network architectures with an unknown number of layers. To relate performance data gathered for different architectures, we define a new kernel for conditional parameter spaces that explicitly includes information about which parameters are relevant in a given structure. We show that this kernel improves model quality and Bayesian optimization results over several simpler baseline kernels.