 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDe novo PROTAC design using graph-based deep generative models
Nov 04, 2022PROteolysis TArgeting Chimeras (PROTACs) are an emerging therapeutic modality for degrading a protein of interest (POI) by marking it for degradation by the proteasome. Recent developments in artificial intelligence (AI) suggest that deep generative models can assist with the de novo design of molecules with desired properties, and their application to PROTAC design remains largely unexplored. We show that a graph-based generative model can be used to propose novel PROTAC-like structures from empty graphs. Our model can be guided towards the generation of large molecules (30--140 heavy atoms) predicted to degrade a POI through policy-gradient reinforcement learning (RL). Rewards during RL are applied using a boosted tree surrogate model that predicts a molecule's degradation potential for each POI. Using this approach, we steer the generative model towards compounds with higher likelihoods of predicted degradation activity. Despite being trained on sparse public data, the generative model proposes molecules with substructures found in known degraders. After fine-tuning, predicted activity against a challenging POI increases from 50% to >80% with near-perfect chemical validity for sampled compounds, suggesting this is a promising approach for the optimization of large, PROTAC-like molecules for targeted protein degradation.
Computer-Aided Multi-Objective Optimization in Small Molecule Discovery
Oct 13, 2022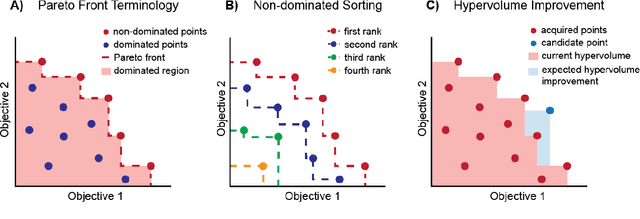
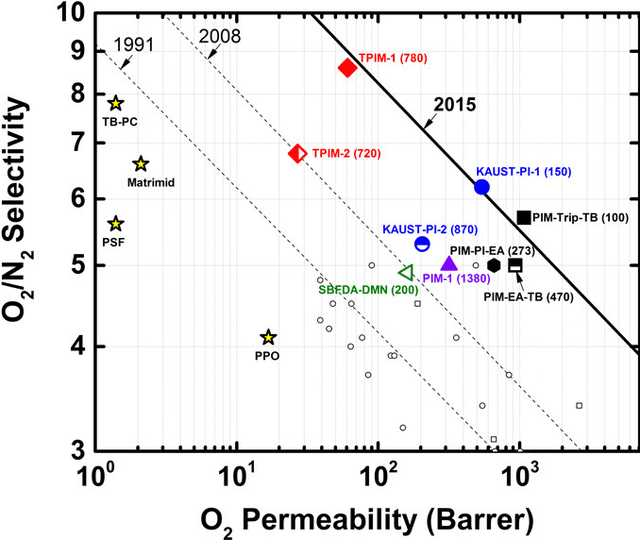
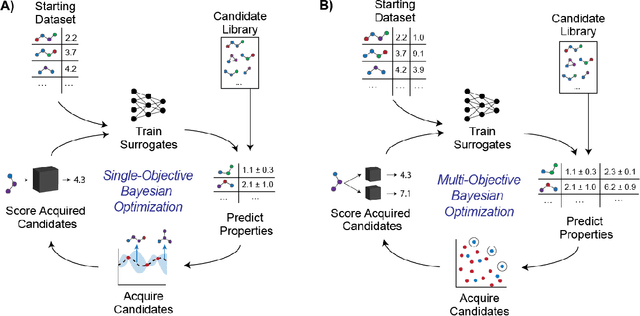
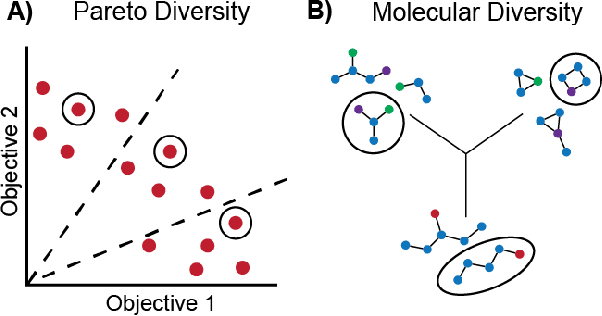
Molecular discovery is a multi-objective optimization problem that requires identifying a molecule or set of molecules that balance multiple, often competing, properties. Multi-objective molecular design is commonly addressed by combining properties of interest into a single objective function using scalarization, which imposes assumptions about relative importance and uncovers little about the trade-offs between objectives. In contrast to scalarization, Pareto optimization does not require knowledge of relative importance and reveals the trade-offs between objectives. However, it introduces additional considerations in algorithm design. In this review, we describe pool-based and de novo generative approaches to multi-objective molecular discovery with a focus on Pareto optimization algorithms. We show how pool-based molecular discovery is a relatively direct extension of multi-objective Bayesian optimization and how the plethora of different generative models extend from single-objective to multi-objective optimization in similar ways using non-dominated sorting in the reward function (reinforcement learning) or to select molecules for retraining (distribution learning) or propagation (genetic algorithms). Finally, we discuss some remaining challenges and opportunities in the field, emphasizing the opportunity to adopt Bayesian optimization techniques into multi-objective de novo design.
Equivariant Shape-Conditioned Generation of 3D Molecules for Ligand-Based Drug Design
Oct 06, 2022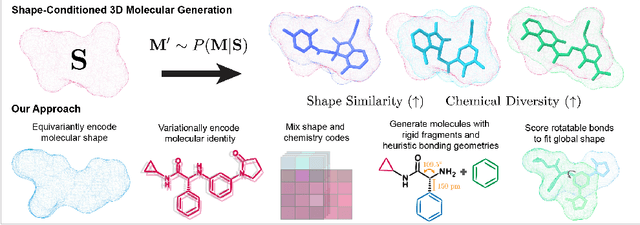
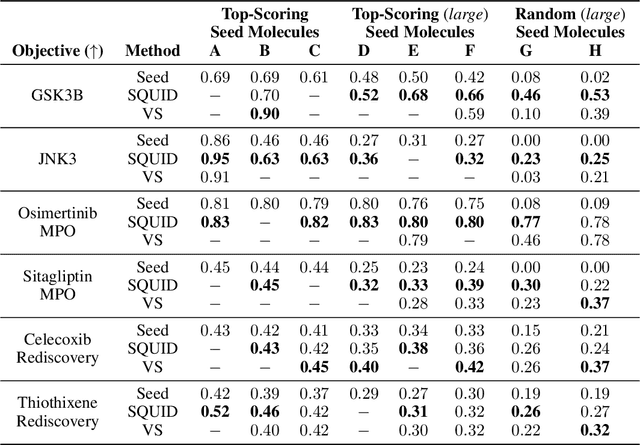
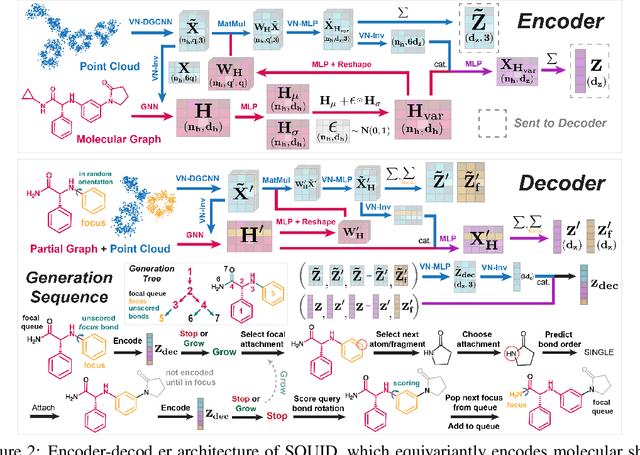

Shape-based virtual screening is widely employed in ligand-based drug design to search chemical libraries for molecules with similar 3D shapes yet novel 2D chemical structures compared to known ligands. 3D deep generative models have the potential to automate this exploration of shape-conditioned 3D chemical space; however, no existing models can reliably generate valid drug-like molecules in conformations that adopt a specific shape such as a known binding pose. We introduce a new multimodal 3D generative model that enables shape-conditioned 3D molecular design by equivariantly encoding molecular shape and variationally encoding chemical identity. We ensure local geometric and chemical validity of generated molecules by using autoregressive fragment-based generation with heuristic bonding geometries, allowing the model to prioritize the scoring of rotatable bonds to best align the growing conformational structure to the target shape. We evaluate our 3D generative model in tasks relevant to drug design including shape-conditioned generation of chemically diverse molecular structures and shape-constrained molecular property optimization, demonstrating its utility over virtual screening of enumerated libraries.
Robust Molecular Image Recognition: A Graph Generation Approach
May 28, 2022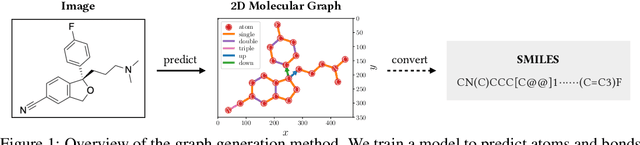
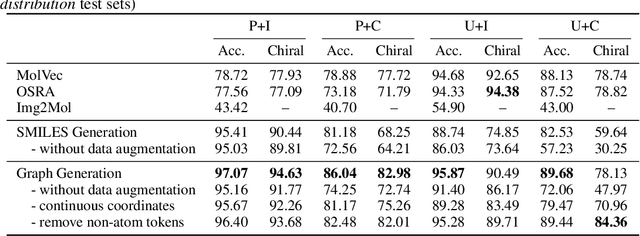
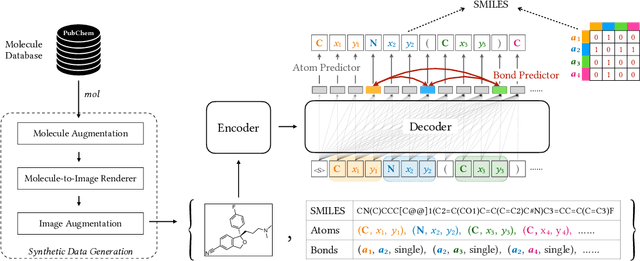
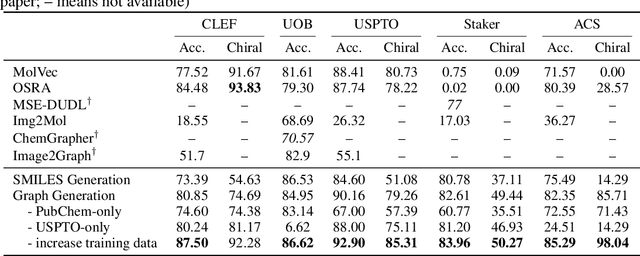
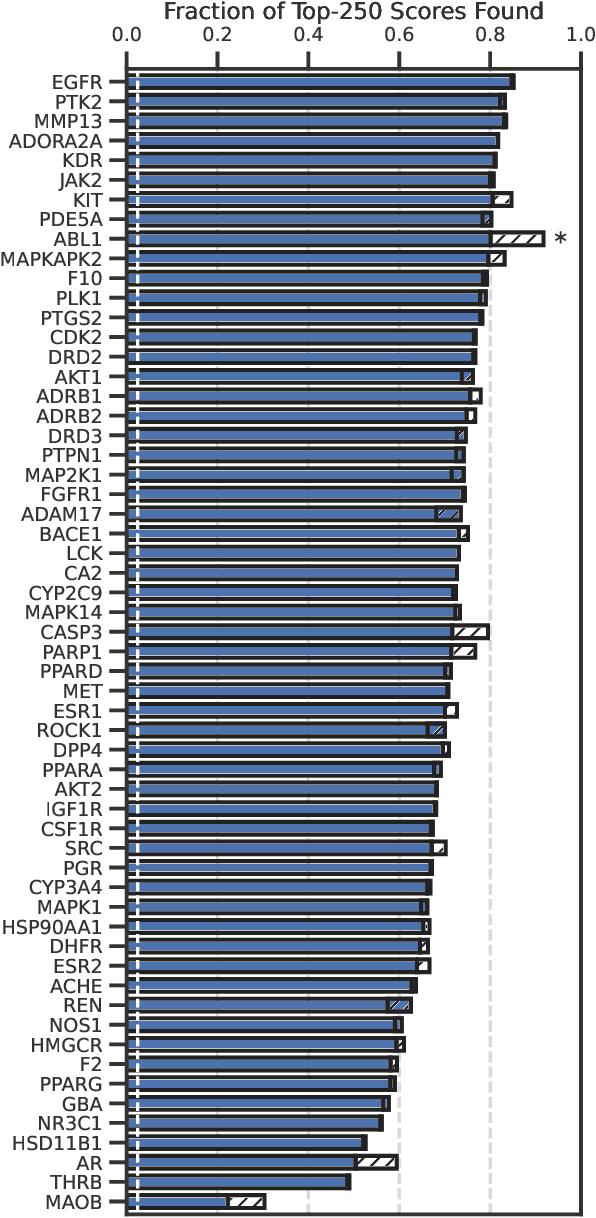
Molecular image recognition is a fundamental task in information extraction from chemistry literature. Previous data-driven models formulate it as an image-to-sequence task, to generate a sequential representation of the molecule (e.g. SMILES string) from its graphical representation. Although they perform adequately on certain benchmarks, these models are not robust in real-world situations, where molecular images differ in style, quality, and chemical patterns. In this paper, we propose a novel graph generation approach that explicitly predicts atoms and bonds, along with their geometric layouts, to construct the molecular graph. We develop data augmentation strategies for molecules and images to increase the robustness of our model against domain shifts. Our model is flexible to incorporate chemistry constraints, and produces more interpretable predictions than SMILES. In experiments on both synthetic and realistic molecular images, our model significantly outperforms previous models, achieving 84-93% accuracy on five benchmarks. We also conduct human evaluation and show that our model reduces the time for a chemist to extract molecular structures from images by roughly 50%.
A graph representation of molecular ensembles for polymer property prediction
May 17, 2022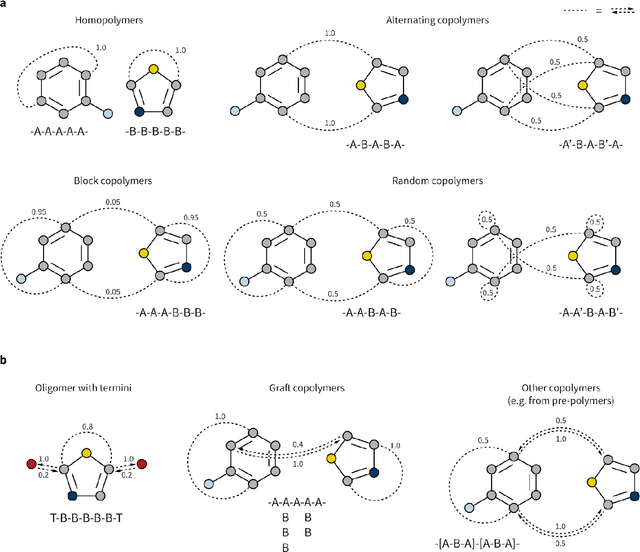
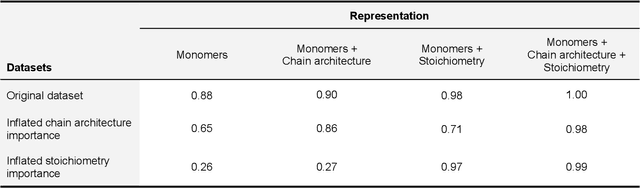
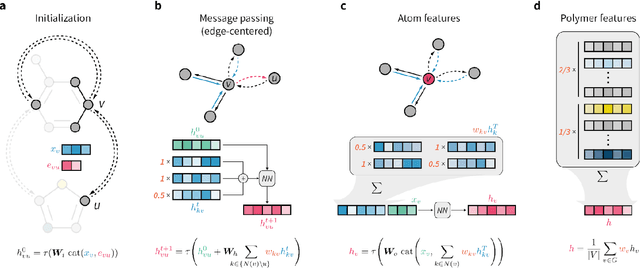
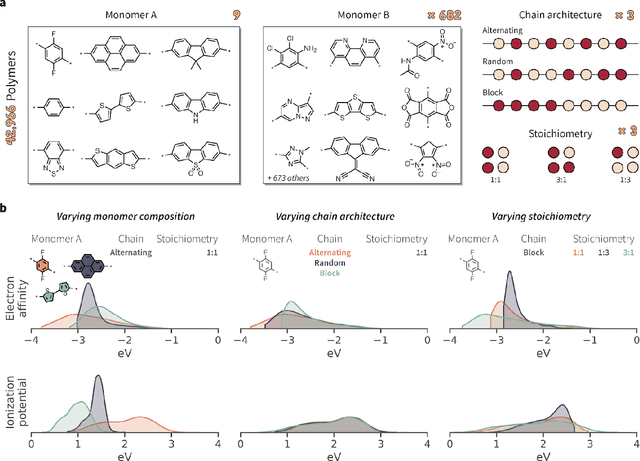
Synthetic polymers are versatile and widely used materials. Similar to small organic molecules, a large chemical space of such materials is hypothetically accessible. Computational property prediction and virtual screening can accelerate polymer design by prioritizing candidates expected to have favorable properties. However, in contrast to organic molecules, polymers are often not well-defined single structures but an ensemble of similar molecules, which poses unique challenges to traditional chemical representations and machine learning approaches. Here, we introduce a graph representation of molecular ensembles and an associated graph neural network architecture that is tailored to polymer property prediction. We demonstrate that this approach captures critical features of polymeric materials, like chain architecture, monomer stoichiometry, and degree of polymerization, and achieves superior accuracy to off-the-shelf cheminformatics methodologies. While doing so, we built a dataset of simulated electron affinity and ionization potential values for >40k polymers with varying monomer composition, stoichiometry, and chain architecture, which may be used in the development of other tailored machine learning approaches. The dataset and machine learning models presented in this work pave the path toward new classes of algorithms for polymer informatics and, more broadly, introduce a framework for the modeling of molecular ensembles.
Self-focusing virtual screening with active design space pruning
May 03, 2022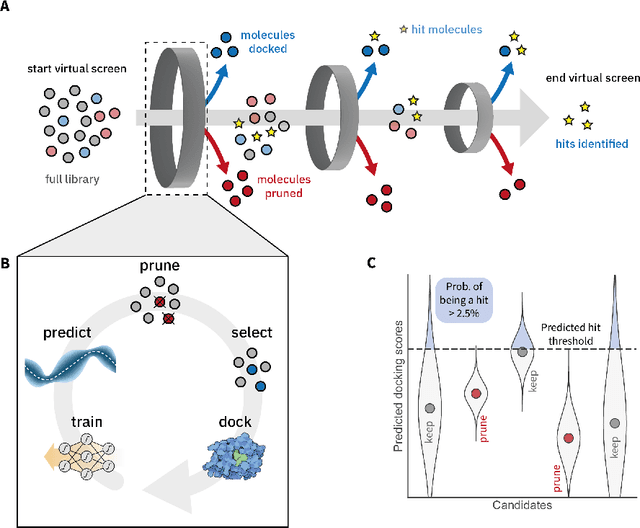
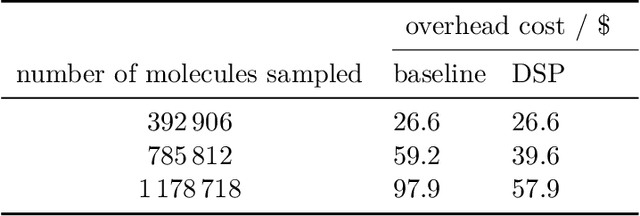
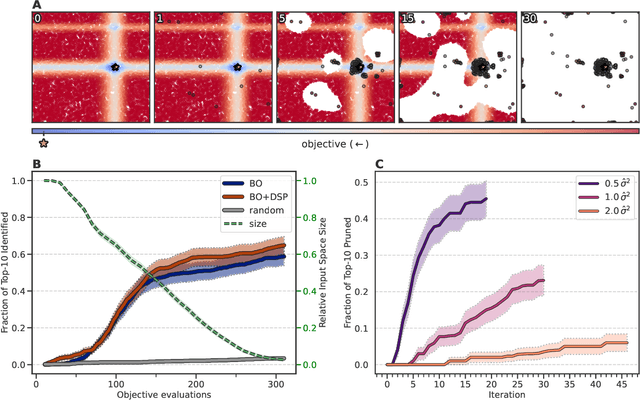
High-throughput virtual screening is an indispensable technique utilized in the discovery of small molecules. In cases where the library of molecules is exceedingly large, the cost of an exhaustive virtual screen may be prohibitive. Model-guided optimization has been employed to lower these costs through dramatic increases in sample efficiency compared to random selection. However, these techniques introduce new costs to the workflow through the surrogate model training and inference steps. In this study, we propose an extension to the framework of model-guided optimization that mitigates inferences costs using a technique we refer to as design space pruning (DSP), which irreversibly removes poor-performing candidates from consideration. We study the application of DSP to a variety of optimization tasks and observe significant reductions in overhead costs while exhibiting similar performance to the baseline optimization. DSP represents an attractive extension of model-guided optimization that can limit overhead costs in optimization settings where these costs are non-negligible relative to objective costs, such as docking.
Bringing Atomistic Deep Learning to Prime Time
Dec 09, 2021
Artificial intelligence has not yet revolutionized the design of materials and molecules. In this perspective, we identify four barriers preventing the integration of atomistic deep learning, molecular science, and high-performance computing. We outline focused research efforts to address the opportunities presented by these challenges.
Scalable Geometric Deep Learning on Molecular Graphs
Dec 06, 2021

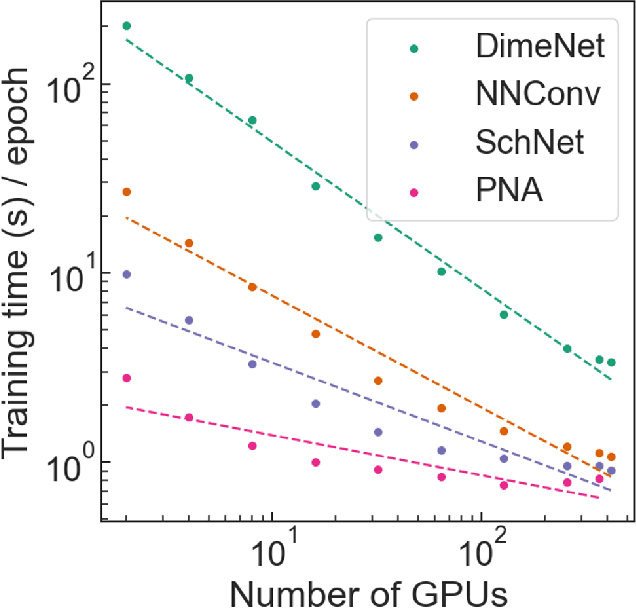
Deep learning in molecular and materials sciences is limited by the lack of integration between applied science, artificial intelligence, and high-performance computing. Bottlenecks with respect to the amount of training data, the size and complexity of model architectures, and the scale of the compute infrastructure are all key factors limiting the scaling of deep learning for molecules and materials. Here, we present $\textit{LitMatter}$, a lightweight framework for scaling molecular deep learning methods. We train four graph neural network architectures on over 400 GPUs and investigate the scaling behavior of these methods. Depending on the model architecture, training time speedups up to $60\times$ are seen. Empirical neural scaling relations quantify the model-dependent scaling and enable optimal compute resource allocation and the identification of scalable molecular geometric deep learning model implementations.
Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction
Oct 19, 2021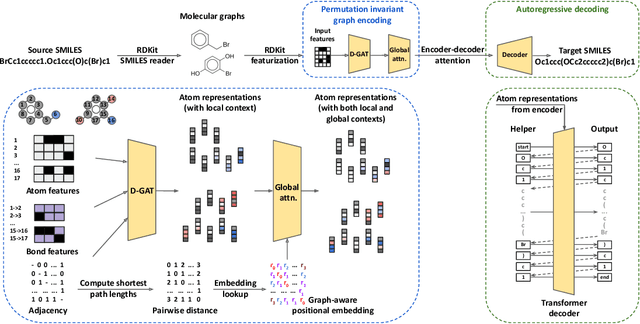
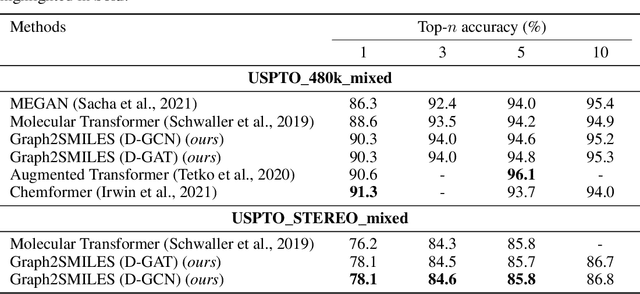
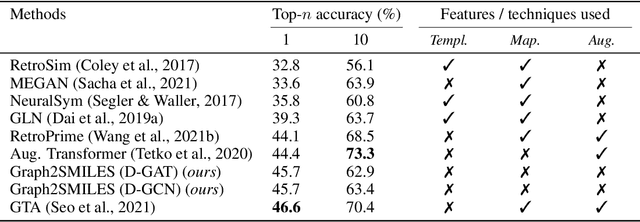
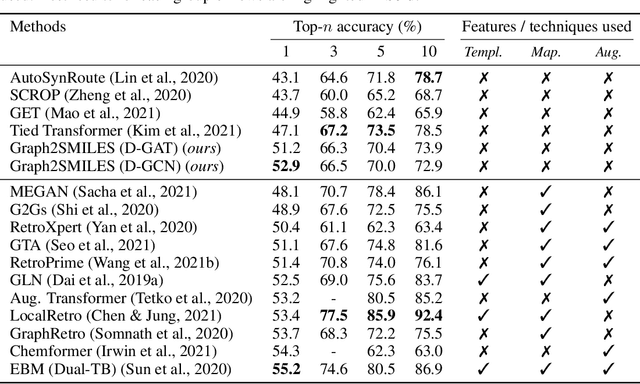
Synthesis planning and reaction outcome prediction are two fundamental problems in computer-aided organic chemistry for which a variety of data-driven approaches have emerged. Natural language approaches that model each problem as a SMILES-to-SMILES translation lead to a simple end-to-end formulation, reduce the need for data preprocessing, and enable the use of well-optimized machine translation model architectures. However, SMILES representations are not an efficient representation for capturing information about molecular structures, as evidenced by the success of SMILES augmentation to boost empirical performance. Here, we describe a novel Graph2SMILES model that combines the power of Transformer models for text generation with the permutation invariance of molecular graph encoders that mitigates the need for input data augmentation. As an end-to-end architecture, Graph2SMILES can be used as a drop-in replacement for the Transformer in any task involving molecule(s)-to-molecule(s) transformations. In our encoder, an attention-augmented directed message passing neural network (D-MPNN) captures local chemical environments, and the global attention encoder allows for long-range and intermolecular interactions, enhanced by graph-aware positional embedding. Graph2SMILES improves the top-1 accuracy of the Transformer baselines by $1.7\%$ and $1.9\%$ for reaction outcome prediction on USPTO_480k and USPTO_STEREO datasets respectively, and by $9.8\%$ for one-step retrosynthesis on the USPTO_50k dataset.
Amortized Tree Generation for Bottom-up Synthesis Planning and Synthesizable Molecular Design
Oct 12, 2021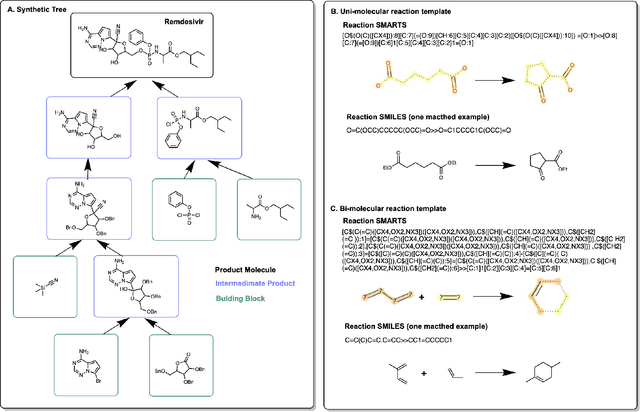
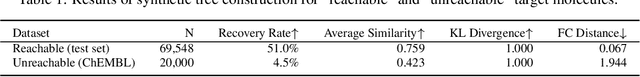
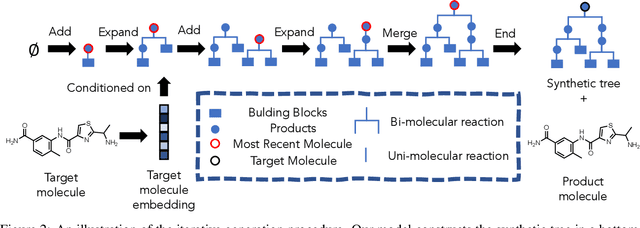
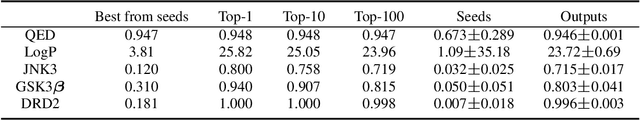
Molecular design and synthesis planning are two critical steps in the process of molecular discovery that we propose to formulate as a single shared task of conditional synthetic pathway generation. We report an amortized approach to generate synthetic pathways as a Markov decision process conditioned on a target molecular embedding. This approach allows us to conduct synthesis planning in a bottom-up manner and design synthesizable molecules by decoding from optimized conditional codes, demonstrating the potential to solve both problems of design and synthesis simultaneously. The approach leverages neural networks to probabilistically model the synthetic trees, one reaction step at a time, according to reactivity rules encoded in a discrete action space of reaction templates. We train these networks on hundreds of thousands of artificial pathways generated from a pool of purchasable compounds and a list of expert-curated templates. We validate our method with (a) the recovery of molecules using conditional generation, (b) the identification of synthesizable structural analogs, and (c) the optimization of molecular structures given oracle functions relevant to drug discovery.