 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentic Personas for Adaptive Scientific Explanations with Knowledge Graphs
Mar 23, 2026AI explanation methods often assume a static user model, producing non-adaptive explanations regardless of expert goals, reasoning strategies, or decision contexts. Knowledge graph-based explanations, despite their capacity for grounded, path-based reasoning, inherit this limitation. In complex domains such as scientific discovery, this assumption fails to capture the diversity of cognitive strategies and epistemic stances among experts, preventing explanations that foster deeper understanding and informed decision-making. However, the scarcity of human experts limits the use of direct human feedback to produce adaptive explanations. We present a reinforcement learning approach for scientific explanation generation that incorporates agentic personas, structured representations of expert reasoning strategies, that guide the explanation agent towards specific epistemic preferences. In an evaluation of knowledge graph-based explanations for drug discovery, we tested two personas that capture distinct epistemic stances derived from expert feedback. Results show that persona-driven explanations match state-of-the-art predictive performance while persona preferences closely align with those of their corresponding experts. Adaptive explanations were consistently preferred over non-adaptive baselines (n = 22), and persona-based training reduces feedback requirements by two orders of magnitude. These findings demonstrate how agentic personas enable scalable adaptive explainability for AI systems in complex and high-stakes domains.
Reinforcement-guided generative protein language models enable de novo design of highly diverse AAV capsids
Mar 19, 2026Adeno-associated viral (AAV) vectors are widely used delivery platforms in gene therapy, and the design of improved capsids is key to expanding their therapeutic potential. A central challenge in AAV bioengineering, as in protein design more broadly, is the vast sequence design space relative to the scale of feasible experimental screening. Machine-guided generative approaches provide a powerful means of navigating this landscape and proposing novel protein sequences that satisfy functional constraints. Here, we develop a generative design framework based on protein language models and reinforcement learning to generate highly novel yet functionally plausible AAV capsids. A pretrained model was fine-tuned on experimentally validated capsid sequences to learn patterns associated with viability. Reinforcement learning was then used to guide sequence generation, with a reward function that jointly promoted predicted viability and sequence novelty, thereby enabling exploration beyond regions represented in the training data. Comparative analyses showed that fine-tuning alone produces sequences with high predicted viability but remains biased toward the training distribution, whereas reinforcement learining-guided generation reaches more distant regions of sequence space while maintaining high predicted viability. Finally, we propose a candidate selection strategy that integrates predicted viability, sequence novelty, and biophysical properties to prioritize variants for downstream evaluation. This work establishes a framework for the generative exploration of protein sequence space and advances the application of generative protein language models to AAV bioengineering.
Exploring the limits of pre-trained embeddings in machine-guided protein design: a case study on predicting AAV vector viability
Feb 16, 2026Effective representations of protein sequences are widely recognized as a cornerstone of machine learning-based protein design. Yet, protein bioengineering poses unique challenges for sequence representation, as experimental datasets typically feature few mutations, which are either sparsely distributed across the entire sequence or densely concentrated within localized regions. This limits the ability of sequence-level representations to extract functionally meaningful signals. In addition, comprehensive comparative studies remain scarce, despite their crucial role in clarifying which representations best encode relevant information and ultimately support superior predictive performance. In this study, we systematically evaluate multiple ProtBERT and ESM2 embedding variants as sequence representations, using the adeno-associated virus capsid as a case study and prototypical example of bioengineering, where functional optimization is targeted through highly localized sequence variation within an otherwise large protein. Our results reveal that, prior to fine-tuning, amino acid-level embeddings outperform sequence-level representations in supervised predictive tasks, whereas the latter tend to be more effective in unsupervised settings. However, optimal performance is only achieved when embeddings are fine-tuned with task-specific labels, with sequence-level representations providing the best performance. Moreover, our findings indicate that the extent of sequence variation required to produce notable shifts in sequence representations exceeds what is typically explored in bioengineering studies, showing the need for fine-tuning in datasets characterized by sparse or highly localized mutations.
CMOMgen: Complex Multi-Ontology Alignment via Pattern-Guided In-Context Learning
Oct 24, 2025



Constructing comprehensive knowledge graphs requires the use of multiple ontologies in order to fully contextualize data into a domain. Ontology matching finds equivalences between concepts interconnecting ontologies and creating a cohesive semantic layer. While the simple pairwise state of the art is well established, simple equivalence mappings cannot provide full semantic integration of related but disjoint ontologies. Complex multi-ontology matching (CMOM) aligns one source entity to composite logical expressions of multiple target entities, establishing more nuanced equivalences and provenance along the ontological hierarchy. We present CMOMgen, the first end-to-end CMOM strategy that generates complete and semantically sound mappings, without establishing any restrictions on the number of target ontologies or entities. Retrieval-Augmented Generation selects relevant classes to compose the mapping and filters matching reference mappings to serve as examples, enhancing In-Context Learning. The strategy was evaluated in three biomedical tasks with partial reference alignments. CMOMgen outperforms baselines in class selection, demonstrating the impact of having a dedicated strategy. Our strategy also achieves a minimum of 63% in F1-score, outperforming all baselines and ablated versions in two out of three tasks and placing second in the third. Furthermore, a manual evaluation of non-reference mappings showed that 46% of the mappings achieve the maximum score, further substantiating its ability to construct semantically sound mappings.
What can knowledge graph alignment gain with Neuro-Symbolic learning approaches?
Oct 11, 2023
Knowledge Graphs (KG) are the backbone of many data-intensive applications since they can represent data coupled with its meaning and context. Aligning KGs across different domains and providers is necessary to afford a fuller and integrated representation. A severe limitation of current KG alignment (KGA) algorithms is that they fail to articulate logical thinking and reasoning with lexical, structural, and semantic data learning. Deep learning models are increasingly popular for KGA inspired by their good performance in other tasks, but they suffer from limitations in explainability, reasoning, and data efficiency. Hybrid neurosymbolic learning models hold the promise of integrating logical and data perspectives to produce high-quality alignments that are explainable and support validation through human-centric approaches. This paper examines the current state of the art in KGA and explores the potential for neurosymbolic integration, highlighting promising research directions for combining these fields.
Knowledge Graphs for the Life Sciences: Recent Developments, Challenges and Opportunities
Oct 06, 2023
The term life sciences refers to the disciplines that study living organisms and life processes, and include chemistry, biology, medicine, and a range of other related disciplines. Research efforts in life sciences are heavily data-driven, as they produce and consume vast amounts of scientific data, much of which is intrinsically relational and graph-structured. The volume of data and the complexity of scientific concepts and relations referred to therein promote the application of advanced knowledge-driven technologies for managing and interpreting data, with the ultimate aim to advance scientific discovery. In this survey and position paper, we discuss recent developments and advances in the use of graph-based technologies in life sciences and set out a vision for how these technologies will impact these fields into the future. We focus on three broad topics: the construction and management of Knowledge Graphs (KGs), the use of KGs and associated technologies in the discovery of new knowledge, and the use of KGs in artificial intelligence applications to support explanations (explainable AI). We select a few exemplary use cases for each topic, discuss the challenges and open research questions within these topics, and conclude with a perspective and outlook that summarizes the overarching challenges and their potential solutions as a guide for future research.
Biomedical Knowledge Graph Embeddings with Negative Statements
Aug 07, 2023



A knowledge graph is a powerful representation of real-world entities and their relations. The vast majority of these relations are defined as positive statements, but the importance of negative statements is increasingly recognized, especially under an Open World Assumption. Explicitly considering negative statements has been shown to improve performance on tasks such as entity summarization and question answering or domain-specific tasks such as protein function prediction. However, no attention has been given to the exploration of negative statements by knowledge graph embedding approaches despite the potential of negative statements to produce more accurate representations of entities in a knowledge graph. We propose a novel approach, TrueWalks, to incorporate negative statements into the knowledge graph representation learning process. In particular, we present a novel walk-generation method that is able to not only differentiate between positive and negative statements but also take into account the semantic implications of negation in ontology-rich knowledge graphs. This is of particular importance for applications in the biomedical domain, where the inadequacy of embedding approaches regarding negative statements at the ontology level has been identified as a crucial limitation. We evaluate TrueWalks in ontology-rich biomedical knowledge graphs in two different predictive tasks based on KG embeddings: protein-protein interaction prediction and gene-disease association prediction. We conduct an extensive analysis over established benchmarks and demonstrate that our method is able to improve the performance of knowledge graph embeddings on all tasks.
Benchmark datasets for biomedical knowledge graphs with negative statements
Jul 21, 2023



Knowledge graphs represent facts about real-world entities. Most of these facts are defined as positive statements. The negative statements are scarce but highly relevant under the open-world assumption. Furthermore, they have been demonstrated to improve the performance of several applications, namely in the biomedical domain. However, no benchmark dataset supports the evaluation of the methods that consider these negative statements. We present a collection of datasets for three relation prediction tasks - protein-protein interaction prediction, gene-disease association prediction and disease prediction - that aim at circumventing the difficulties in building benchmarks for knowledge graphs with negative statements. These datasets include data from two successful biomedical ontologies, Gene Ontology and Human Phenotype Ontology, enriched with negative statements. We also generate knowledge graph embeddings for each dataset with two popular path-based methods and evaluate the performance in each task. The results show that the negative statements can improve the performance of knowledge graph embeddings.
Explainable Representations for Relation Prediction in Knowledge Graphs
Jun 22, 2023Knowledge graphs represent real-world entities and their relations in a semantically-rich structure supported by ontologies. Exploring this data with machine learning methods often relies on knowledge graph embeddings, which produce latent representations of entities that preserve structural and local graph neighbourhood properties, but sacrifice explainability. However, in tasks such as link or relation prediction, understanding which specific features better explain a relation is crucial to support complex or critical applications. We propose SEEK, a novel approach for explainable representations to support relation prediction in knowledge graphs. It is based on identifying relevant shared semantic aspects (i.e., subgraphs) between entities and learning representations for each subgraph, producing a multi-faceted and explainable representation. We evaluate SEEK on two real-world highly complex relation prediction tasks: protein-protein interaction prediction and gene-disease association prediction. Our extensive analysis using established benchmarks demonstrates that SEEK achieves significantly better performance than standard learning representation methods while identifying both sufficient and necessary explanations based on shared semantic aspects.
Predicting Gene-Disease Associations with Knowledge Graph Embeddings over Multiple Ontologies
May 31, 2021
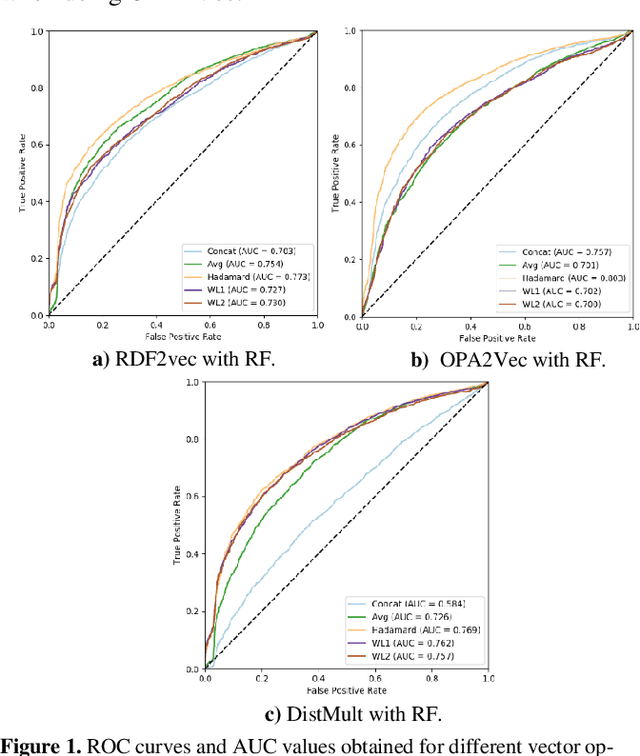
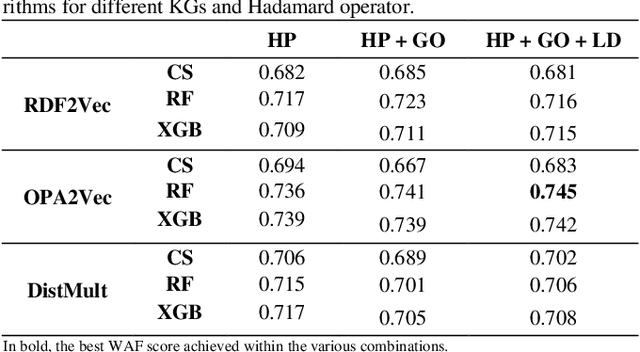
Ontology-based approaches for predicting gene-disease associations include the more classical semantic similarity methods and more recently knowledge graph embeddings. While semantic similarity is typically restricted to hierarchical relations within the ontology, knowledge graph embeddings consider their full breadth. However, embeddings are produced over a single graph and complex tasks such as gene-disease association may require additional ontologies. We investigate the impact of employing richer semantic representations that are based on more than one ontology, able to represent both genes and diseases and consider multiple kinds of relations within the ontologies. Our experiments demonstrate the value of employing knowledge graph embeddings based on random-walks and highlight the need for a closer integration of different ontologies.