 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePyTrial: A Comprehensive Platform for Artificial Intelligence for Drug Development
Jun 06, 2023



Drug development is a complex process that aims to test the efficacy and safety of candidate drugs in the human body for regulatory approval via clinical trials. Recently, machine learning has emerged as a vital tool for drug development, offering new opportunities to improve the efficiency and success rates of the process. To facilitate the research and development of artificial intelligence (AI) for drug development, we developed a Python package, namely PyTrial, that implements various clinical trial tasks supported by AI algorithms. To be specific, PyTrial implements 6 essential drug development tasks, including patient outcome prediction, trial site selection, trial outcome prediction, patient-trial matching, trial similarity search, and synthetic data generation. In PyTrial, all tasks are defined by four steps: load data, model definition, model training, and model evaluation, which can be done with a couple of lines of code. In addition, the modular API design allows practitioners to extend the framework to new algorithms and tasks easily. PyTrial is featured for a unified API, detailed documentation, and interactive examples with preprocessed benchmark data for all implemented algorithms. This package can be installed through Python Package Index (PyPI) and is publicly available at https://github.com/RyanWangZf/PyTrial.
FRAMM: Fair Ranking with Missing Modalities for Clinical Trial Site Selection
May 30, 2023



Despite many efforts to address the disparities, the underrepresentation of gender, racial, and ethnic minorities in clinical trials remains a problem and undermines the efficacy of treatments on minorities. This paper focuses on the trial site selection task and proposes FRAMM, a deep reinforcement learning framework for fair trial site selection. We focus on addressing two real-world challenges that affect fair trial sites selection: the data modalities are often not complete for many potential trial sites, and the site selection needs to simultaneously optimize for both enrollment and diversity since the problem is necessarily a trade-off between the two with the only possible way to increase diversity post-selection being through limiting enrollment via caps. To address the missing data challenge, FRAMM has a modality encoder with a masked cross-attention mechanism for handling missing data, bypassing data imputation and the need for complete data in training. To handle the need for making efficient trade-offs, FRAMM uses deep reinforcement learning with a specifically designed reward function that simultaneously optimizes for both enrollment and fairness. We evaluate FRAMM using 4,392 real-world clinical trials ranging from 2016 to 2021 and show that FRAMM outperforms the leading baseline in enrollment-only settings while also achieving large gains in diversity. Specifically, it is able to produce a 9% improvement in diversity with similar enrollment levels over the leading baselines. That improved diversity is further manifested in achieving up to a 14% increase in Hispanic enrollment, 27% increase in Black enrollment, and 60% increase in Asian enrollment compared to selecting sites with an enrollment-only model.
GraphCare: Enhancing Healthcare Predictions with Open-World Personalized Knowledge Graphs
May 22, 2023Clinical predictive models often rely on patients electronic health records (EHR), but integrating medical knowledge to enhance predictions and decision-making is challenging. This is because personalized predictions require personalized knowledge graphs (KGs), which are difficult to generate from patient EHR data. To address this, we propose GraphCare, an open-world framework that leverages external KGs to improve EHR-based predictions. Our method extracts knowledge from large language models (LLMs) and external biomedical KGs to generate patient-specific KGs, which are then used to train our proposed Bi-attention AugmenTed BAT graph neural network GNN for healthcare predictions. We evaluate GraphCare on two public datasets: MIMIC-III and MIMIC-IV. Our method outperforms baseline models in four vital healthcare prediction tasks: mortality, readmission, length-of-stay, and drug recommendation, improving AUROC on MIMIC-III by average margins of 10.4%, 3.8%, 2.0%, and 1.5%, respectively. Notably, GraphCare demonstrates a substantial edge in scenarios with limited data availability. Our findings highlight the potential of using external KGs in healthcare prediction tasks and demonstrate the promise of GraphCare in generating personalized KGs for promoting personalized medicine.
AnyPredict: Foundation Model for Tabular Prediction
May 20, 2023



Foundation models are pre-trained on massive data to perform well across many downstream tasks. They have demonstrated significant success in natural language processing and computer vision. Nonetheless, the use of such models in tabular prediction tasks has been limited, with the main hurdles consisting of (1) the lack of large-scale and diverse tabular datasets with standardized labels and (2) the schema mismatch and predictive target heterogeneity across domains. This paper proposes a method for building training data at scale for tabular prediction foundation models (AnyPredict) using both in-domain and a wide range of out-domain datasets. The method uses a data engine that leverages large language models (LLMs) to consolidate tabular samples to overcome the barrier across tables with varying schema and align out-domain data with the target task using a ``learn, annotate, and audit'' pipeline. The expanded training data enables the pre-trained AnyPredict to support every tabular dataset in the domain without fine-tuning, resulting in significant improvements over supervised baselines: it reaches an average ranking of 1.57 and 1.00 on 7 patient outcome prediction datasets and 3 trial outcome prediction datasets, respectively. In addition, AnyPredict exhibits impressive zero-shot performances: it outperforms supervised XGBoost models by 8.9% and 17.2% on average in two prediction tasks, respectively.
AutoTrial: Prompting Language Models for Clinical Trial Design
May 19, 2023



Clinical trials are critical for drug development. Constructing the appropriate eligibility criteria (i.e., the inclusion/exclusion criteria for patient recruitment) is essential for the trial's success. Proper design of clinical trial protocols should consider similar precedent trials and their eligibility criteria to ensure sufficient patient coverage. In this paper, we present a method named AutoTrial to aid the design of clinical eligibility criteria using language models. It allows (1) controllable generation under instructions via a hybrid of discrete and neural prompting, (2) scalable knowledge incorporation via in-context learning, and (3) explicit reasoning chains to provide rationales for understanding the outputs. Experiments on over 70K clinical trials verify that AutoTrial generates high-quality criteria texts that are fluent and coherent and with high accuracy in capturing the relevant clinical concepts to the target trial. It is noteworthy that our method, with a much smaller parameter size, gains around 60\% winning rate against the GPT-3.5 baselines via human evaluations.
SPOT: Sequential Predictive Modeling of Clinical Trial Outcome with Meta-Learning
Apr 07, 2023



Clinical trials are essential to drug development but time-consuming, costly, and prone to failure. Accurate trial outcome prediction based on historical trial data promises better trial investment decisions and more trial success. Existing trial outcome prediction models were not designed to model the relations among similar trials, capture the progression of features and designs of similar trials, or address the skewness of trial data which causes inferior performance for less common trials. To fill the gap and provide accurate trial outcome prediction, we propose Sequential Predictive mOdeling of clinical Trial outcome (SPOT) that first identifies trial topics to cluster the multi-sourced trial data into relevant trial topics. It then generates trial embeddings and organizes them by topic and time to create clinical trial sequences. With the consideration of each trial sequence as a task, it uses a meta-learning strategy to achieve a point where the model can rapidly adapt to new tasks with minimal updates. In particular, the topic discovery module enables a deeper understanding of the underlying structure of the data, while sequential learning captures the evolution of trial designs and outcomes. This results in predictions that are not only more accurate but also more interpretable, taking into account the temporal patterns and unique characteristics of each trial topic. We demonstrate that SPOT wins over the prior methods by a significant margin on trial outcome benchmark data: with a 21.5\% lift on phase I, an 8.9\% lift on phase II, and a 5.5\% lift on phase III trials in the metric of the area under precision-recall curve (PR-AUC).
Synthesize Extremely High-dimensional Longitudinal Electronic Health Records via Hierarchical Autoregressive Language Model
Apr 04, 2023Synthetic electronic health records (EHRs) that are both realistic and preserve privacy can serve as an alternative to real EHRs for machine learning (ML) modeling and statistical analysis. However, generating high-fidelity and granular electronic health record (EHR) data in its original, highly-dimensional form poses challenges for existing methods due to the complexities inherent in high-dimensional data. In this paper, we propose Hierarchical Autoregressive Language mOdel (HALO) for generating longitudinal high-dimensional EHR, which preserve the statistical properties of real EHR and can be used to train accurate ML models without privacy concerns. Our HALO method, designed as a hierarchical autoregressive model, generates a probability density function of medical codes, clinical visits, and patient records, allowing for the generation of realistic EHR data in its original, unaggregated form without the need for variable selection or aggregation. Additionally, our model also produces high-quality continuous variables in a longitudinal and probabilistic manner. We conducted extensive experiments and demonstrate that HALO can generate high-fidelity EHR data with high-dimensional disease code probabilities (d > 10,000), disease co-occurrence probabilities within visits (d > 1,000,000), and conditional probabilities across consecutive visits (d > 5,000,000) and achieve above 0.9 R2 correlation in comparison to real EHR data. This performance then enables downstream ML models trained on its synthetic data to achieve comparable accuracy to models trained on real data (0.938 AUROC with HALO data vs. 0.943 with real data). Finally, using a combination of real and synthetic data enhances the accuracy of ML models beyond that achieved by using only real EHR data.
Fast Online Value-Maximizing Prediction Sets with Conformal Cost Control
Feb 02, 2023Many real-world multi-label prediction problems involve set-valued predictions that must satisfy specific requirements dictated by downstream usage. We focus on a typical scenario where such requirements, separately encoding \textit{value} and \textit{cost}, compete with each other. For instance, a hospital might expect a smart diagnosis system to capture as many severe, often co-morbid, diseases as possible (the value), while maintaining strict control over incorrect predictions (the cost). We present a general pipeline, dubbed as FavMac, to maximize the value while controlling the cost in such scenarios. FavMac can be combined with almost any multi-label classifier, affording distribution-free theoretical guarantees on cost control. Moreover, unlike prior works, FavMac can handle real-world large-scale applications via a carefully designed online update mechanism, which is of independent interest. Our methodological and theoretical contributions are supported by experiments on several healthcare tasks and synthetic datasets - FavMac furnishes higher value compared with several variants and baselines while maintaining strict cost control.
Clinical trial site matching with improved diversity using fair policy learning
Apr 13, 2022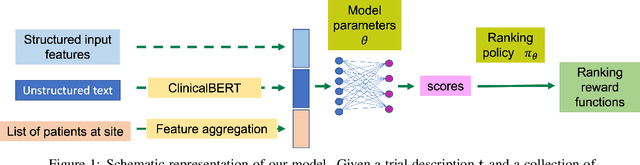
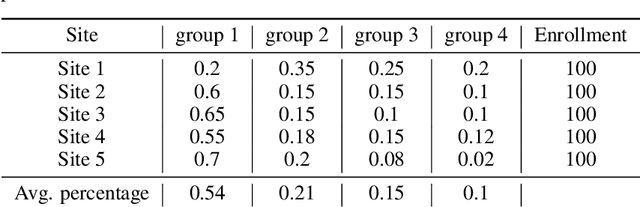
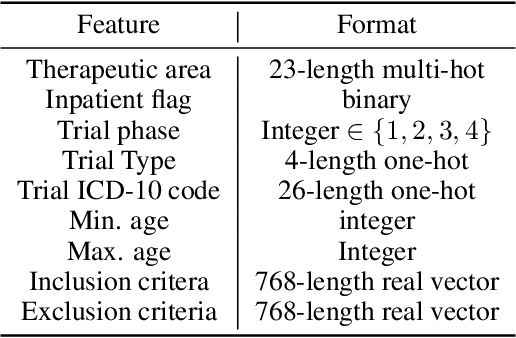
The ongoing pandemic has highlighted the importance of reliable and efficient clinical trials in healthcare. Trial sites, where the trials are conducted, are chosen mainly based on feasibility in terms of medical expertise and access to a large group of patients. More recently, the issue of diversity and inclusion in clinical trials is gaining importance. Different patient groups may experience the effects of a medical drug/ treatment differently and hence need to be included in the clinical trials. These groups could be based on ethnicity, co-morbidities, age, or economic factors. Thus, designing a method for trial site selection that accounts for both feasibility and diversity is a crucial and urgent goal. In this paper, we formulate this problem as a ranking problem with fairness constraints. Using principles of fairness in machine learning, we learn a model that maps a clinical trial description to a ranked list of potential trial sites. Unlike existing fairness frameworks, the group membership of each trial site is non-binary: each trial site may have access to patients from multiple groups. We propose fairness criteria based on demographic parity to address such a multi-group membership scenario. We test our method on 480 real-world clinical trials and show that our model results in a list of potential trial sites that provides access to a diverse set of patients while also ensuing a high number of enrolled patients.
AutoMap: Automatic Medical Code Mapping for Clinical Prediction Model Deployment
Mar 04, 2022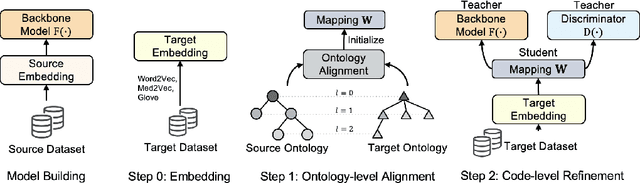
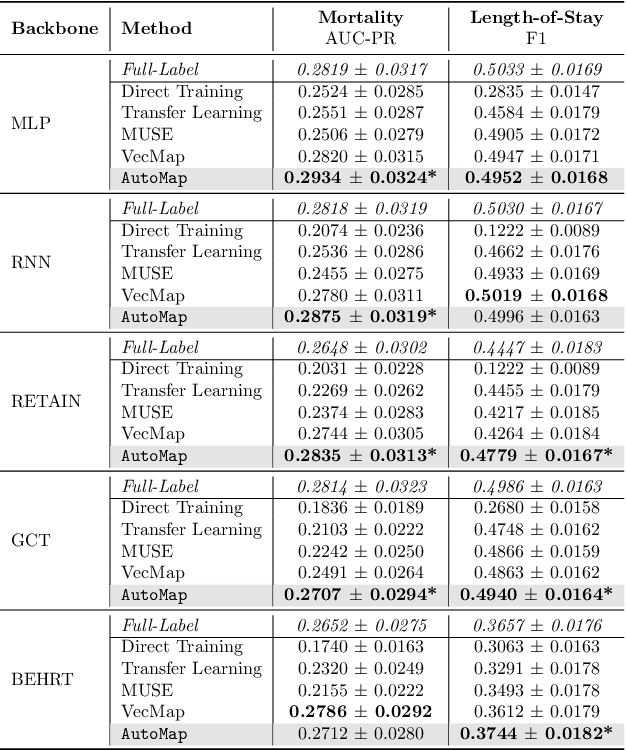
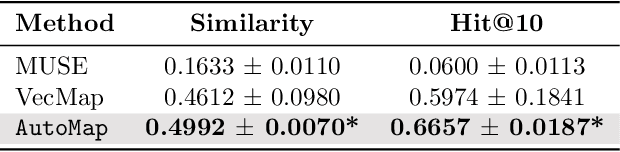
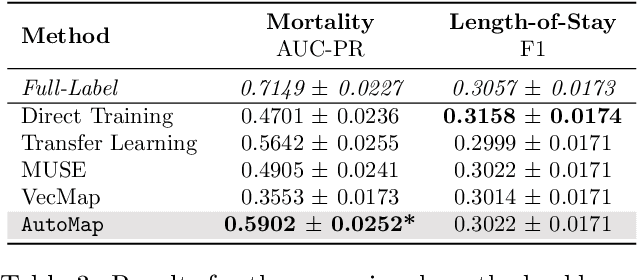
Given a deep learning model trained on data from a source site, how to deploy the model to a target hospital automatically? How to accommodate heterogeneous medical coding systems across different hospitals? Standard approaches rely on existing medical code mapping tools, which have significant practical limitations. To tackle this problem, we propose AutoMap to automatically map the medical codes across different EHR systems in a coarse-to-fine manner: (1) Ontology-level Alignment: We leverage the ontology structure to learn a coarse alignment between the source and target medical coding systems; (2) Code-level Refinement: We refine the alignment at a fine-grained code level for the downstream tasks using a teacher-student framework. We evaluate AutoMap using several deep learning models with two real-world EHR datasets: eICU and MIMIC-III. Results show that AutoMap achieves relative improvements up to 3.9% (AUC-ROC) and 8.7% (AUC-PR) for mortality prediction, and up to 4.7% (AUC-ROC) and 3.7% (F1) for length-of-stay estimation. Further, we show that AutoMap can provide accurate mapping across coding systems. Lastly, we demonstrate that AutoMap can adapt to the two challenging scenarios: (1) mapping between completely different coding systems and (2) between completely different hospitals.