 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBOOM: Benchmarking Out-Of-distribution Molecular Property Predictions of Machine Learning Models
May 03, 2025Advances in deep learning and generative modeling have driven interest in data-driven molecule discovery pipelines, whereby machine learning (ML) models are used to filter and design novel molecules without requiring prohibitively expensive first-principles simulations. Although the discovery of novel molecules that extend the boundaries of known chemistry requires accurate out-of-distribution (OOD) predictions, ML models often struggle to generalize OOD. Furthermore, there are currently no systematic benchmarks for molecular OOD prediction tasks. We present BOOM, $\boldsymbol{b}$enchmarks for $\boldsymbol{o}$ut-$\boldsymbol{o}$f-distribution $\boldsymbol{m}$olecular property predictions -- a benchmark study of property-based out-of-distribution models for common molecular property prediction models. We evaluate more than 140 combinations of models and property prediction tasks to benchmark deep learning models on their OOD performance. Overall, we do not find any existing models that achieve strong OOD generalization across all tasks: even the top performing model exhibited an average OOD error 3x larger than in-distribution. We find that deep learning models with high inductive bias can perform well on OOD tasks with simple, specific properties. Although chemical foundation models with transfer and in-context learning offer a promising solution for limited training data scenarios, we find that current foundation models do not show strong OOD extrapolation capabilities. We perform extensive ablation experiments to highlight how OOD performance is impacted by data generation, pre-training, hyperparameter optimization, model architecture, and molecular representation. We propose that developing ML models with strong OOD generalization is a new frontier challenge in chemical ML model development. This open-source benchmark will be made available on Github.
Active Learning Enables Extrapolation in Molecular Generative Models
Jan 03, 2025



Although generative models hold promise for discovering molecules with optimized desired properties, they often fail to suggest synthesizable molecules that improve upon the known molecules seen in training. We find that a key limitation is not in the molecule generation process itself, but in the poor generalization capabilities of molecular property predictors. We tackle this challenge by creating an active-learning, closed-loop molecule generation pipeline, whereby molecular generative models are iteratively refined on feedback from quantum chemical simulations to improve generalization to new chemical space. Compared against other generative model approaches, only our active learning approach generates molecules with properties that extrapolate beyond the training data (reaching up to 0.44 standard deviations beyond the training data range) and out-of-distribution molecule classification accuracy is improved by 79%. By conditioning molecular generation on thermodynamic stability data from the active-learning loop, the proportion of stable molecules generated is 3.5x higher than the next-best model.
Representing Polymers as Periodic Graphs with Learned Descriptors for Accurate Polymer Property Predictions
May 27, 2022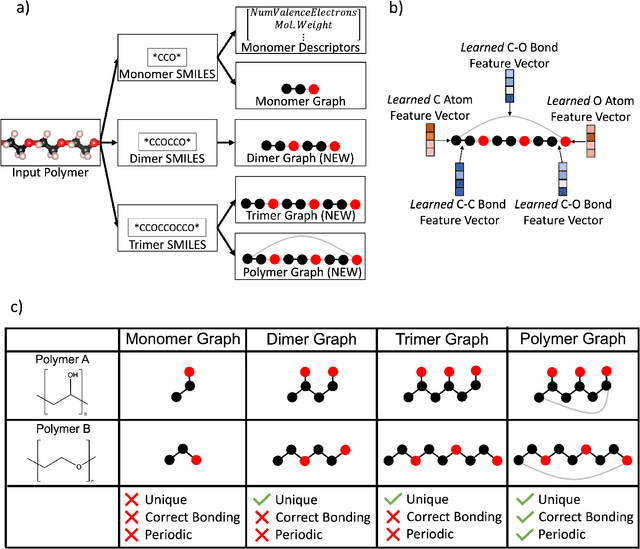
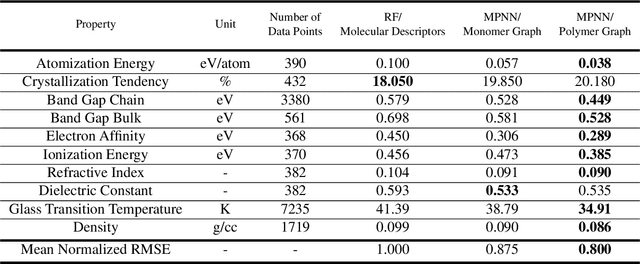
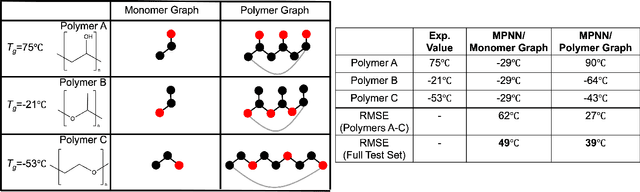
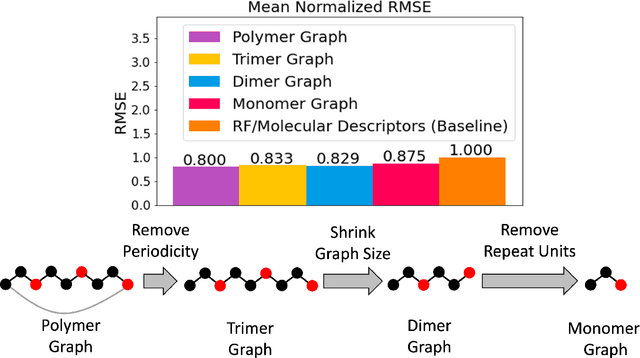
One of the grand challenges of utilizing machine learning for the discovery of innovative new polymers lies in the difficulty of accurately representing the complex structures of polymeric materials. Although a wide array of hand-designed polymer representations have been explored, there has yet to be an ideal solution for how to capture the periodicity of polymer structures, and how to develop polymer descriptors without the need for human feature design. In this work, we tackle these problems through the development of our periodic polymer graph representation. Our pipeline for polymer property predictions is comprised of our polymer graph representation that naturally accounts for the periodicity of polymers, followed by a message-passing neural network (MPNN) that leverages the power of graph deep learning to automatically learn chemically-relevant polymer descriptors. Across a diverse dataset of 10 polymer properties, we find that this polymer graph representation consistently outperforms hand-designed representations with a 20% average reduction in prediction error. Our results illustrate how the incorporation of chemical intuition through directly encoding periodicity into our polymer graph representation leads to a considerable improvement in the accuracy and reliability of polymer property predictions. We also demonstrate how combining polymer graph representations with message-passing neural network architectures can automatically extract meaningful polymer features that are consistent with human intuition, while outperforming human-derived features. This work highlights the advancement in predictive capability that is possible if using chemical descriptors that are specifically optimized for capturing the unique chemical structure of polymers.
Explainable Deep Learning for Uncovering Actionable Scientific Insights for Materials Discovery and Design
Jul 16, 2020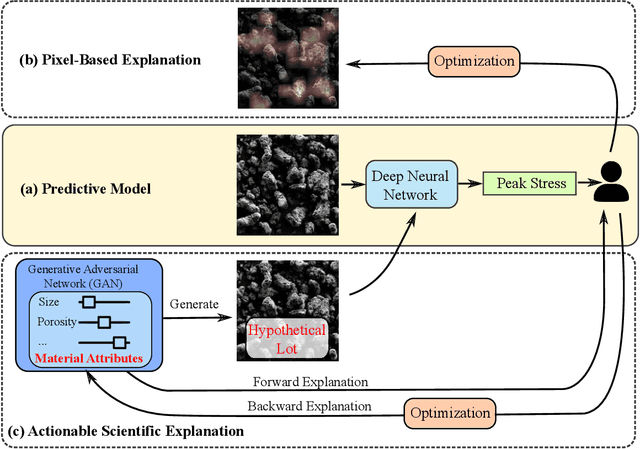


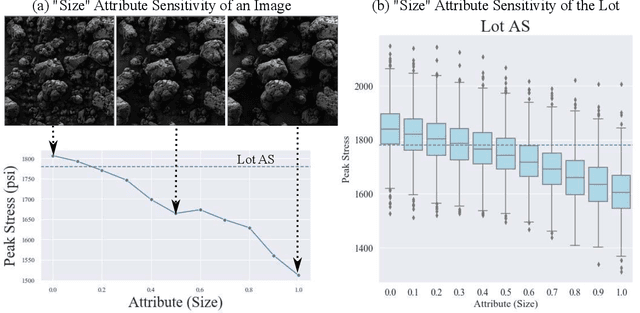
The scientific community has been increasingly interested in harnessing the power of deep learning to solve various domain challenges. However, despite the effectiveness in building predictive models, fundamental challenges exist in extracting actionable knowledge from deep neural networks due to their opaque nature. In this work, we propose techniques for exploring the behavior of deep learning models by injecting domain-specific actionable attributes as tunable "knobs" in the analysis pipeline. By incorporating the domain knowledge in a generative modeling framework, we are not only able to better understand the behavior of these black-box models, but also provide scientists with actionable insights that can potentially lead to fundamental discoveries.
Actionable Attribution Maps for Scientific Machine Learning
Jun 30, 2020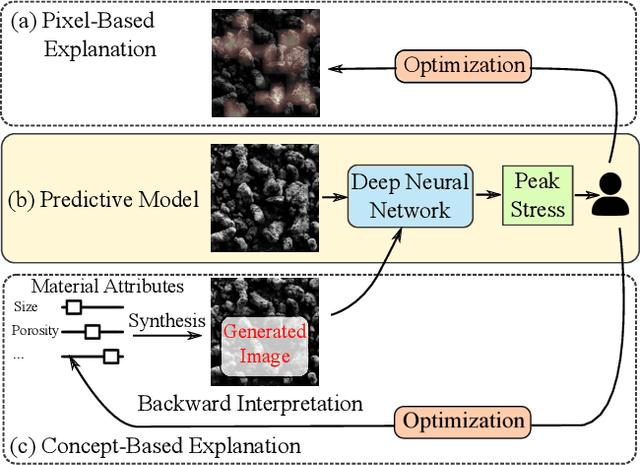

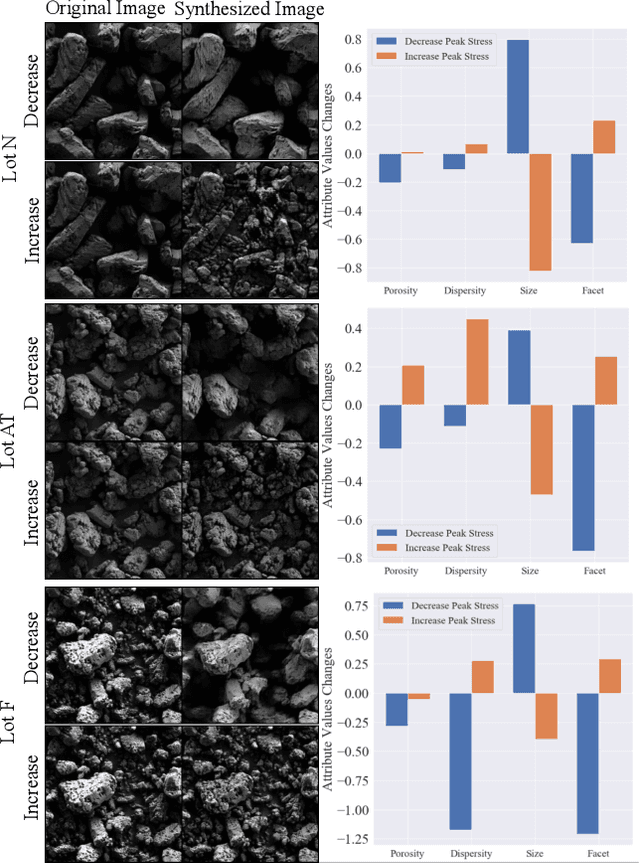
The scientific community has been increasingly interested in harnessing the power of deep learning to solve various domain challenges. However, despite the effectiveness in building predictive models, fundamental challenges exist in extracting actionable knowledge from the deep neural network due to their opaque nature. In this work, we propose techniques for exploring the behavior of deep learning models by injecting domain-specific actionable concepts as tunable ``knobs'' in the analysis pipeline. By incorporating the domain knowledge with generative modeling, we are not only able to better understand the behavior of these black-box models, but also provide scientists with actionable insights that can potentially lead to fundamental discoveries.