 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInverse design of bespoke interatomic potentials via active learning by information-matching
Jun 06, 2026Interatomic potentials (IPs) enable large-scale atomistic simulations beyond the reach of first-principles methods, but their predictive reliability depends critically on the selection of training data, quantified uncertainty, and model expressiveness. Active learning (AL) provides a principled framework for constructing efficient and accurate IPs, yet most strategies reduce parameter uncertainty without explicitly accounting for the specific material properties being predicted. The information-matching (IM) approach addresses this limitation by requiring that the selected training data provide at least as much parameter space information as needed to achieve prescribed uncertainty targets for selected quantities of interest (QoIs). Here, we apply IM to develop bespoke IPs specifically tailored for predicting plastic strength in metals. Due to the high computational cost of simulating plastic strength, we employ an indirect IM strategy that targets inexpensive intermediate QoIs that correlate with strength. The IM method enables precise parameter constraints with minimal training data, yielding precise predictions for both the intermediate QoIs and plastic strength. Yet, model error remains a key limitation, and a post hoc uncertainty inflation correction provides a viable means to mitigate this limitation. These findings illustrate both the promise and limits of uncertainty-aware AL for predicting complex material properties.
Unsupervised Atomic Data Mining via Multi-Kernel Graph Autoencoders for Machine Learning Force Fields
Sep 15, 2025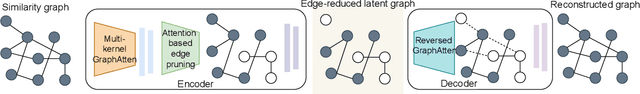
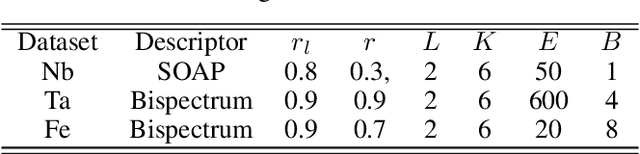
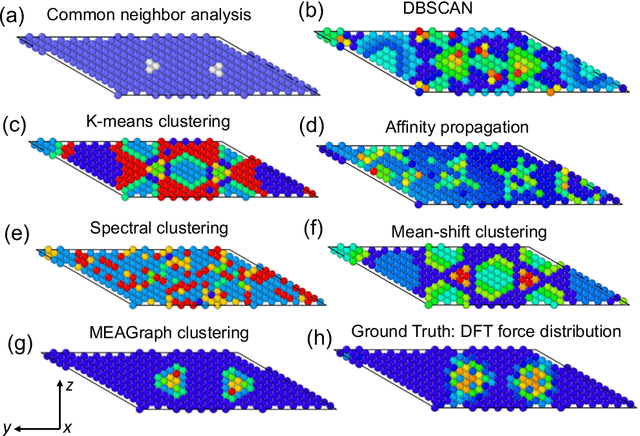
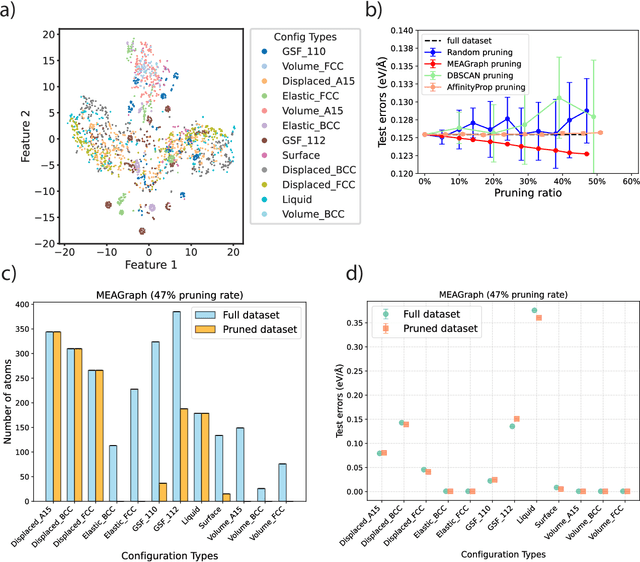
Constructing a chemically diverse dataset while avoiding sampling bias is critical to training efficient and generalizable force fields. However, in computational chemistry and materials science, many common dataset generation techniques are prone to oversampling regions of the potential energy surface. Furthermore, these regions can be difficult to identify and isolate from each other or may not align well with human intuition, making it challenging to systematically remove bias in the dataset. While traditional clustering and pruning (down-sampling) approaches can be useful for this, they can often lead to information loss or a failure to properly identify distinct regions of the potential energy surface due to difficulties associated with the high dimensionality of atomic descriptors. In this work, we introduce the Multi-kernel Edge Attention-based Graph Autoencoder (MEAGraph) model, an unsupervised approach for analyzing atomic datasets. MEAGraph combines multiple linear kernel transformations with attention-based message passing to capture geometric sensitivity and enable effective dataset pruning without relying on labels or extensive training. Demonstrated applications on niobium, tantalum, and iron datasets show that MEAGraph efficiently groups similar atomic environments, allowing for the use of basic pruning techniques for removing sampling bias. This approach provides an effective method for representation learning and clustering that can be used for data analysis, outlier detection, and dataset optimization.
Composable and adaptive design of machine learning interatomic potentials guided by Fisher-information analysis
Apr 27, 2025An adaptive physics-informed model design strategy for machine-learning interatomic potentials (MLIPs) is proposed. This strategy follows an iterative reconfiguration of composite models from single-term models, followed by a unified training procedure. A model evaluation method based on the Fisher information matrix (FIM) and multiple-property error metrics is proposed to guide model reconfiguration and hyperparameter optimization. Combining the model reconfiguration and the model evaluation subroutines, we provide an adaptive MLIP design strategy that balances flexibility and extensibility. In a case study of designing models against a structurally diverse niobium dataset, we managed to obtain an optimal configuration with 75 parameters generated by our framework that achieved a force RMSE of 0.172 eV/{\AA} and an energy RMSE of 0.013 eV/atom.
LTAU-FF: Loss Trajectory Analysis for Uncertainty in Atomistic Force Fields
Feb 01, 2024Model ensembles are simple and effective tools for estimating the prediction uncertainty of deep learning atomistic force fields. Despite this, widespread adoption of ensemble-based uncertainty quantification (UQ) techniques is limited by the high computational costs incurred by ensembles during both training and inference. In this work we leverage the cumulative distribution functions (CDFs) of per-sample errors obtained over the course of training to efficiently represent the model ensemble, and couple them with a distance-based similarity search in the model latent space. Using these tools, we develop a simple UQ metric (which we call LTAU) that leverages the strengths of ensemble-based techniques without requiring the evaluation of multiple models during either training or inference. As an initial test, we apply our method towards estimating the epistemic uncertainty in atomistic force fields (LTAU-FF) and demonstrate that it can be easily calibrated to accurately predict test errors on multiple datasets from the literature. We then illustrate the utility of LTAU-FF in two practical applications: 1) tuning the training-validation gap for an example dataset, and 2) predicting errors in relaxation trajectories on the OC20 IS2RS task. Though in this work we focus on the use of LTAU with deep learning atomistic force fields, we emphasize that it can be readily applied to any regression task, or any ensemble-generation technique, to provide a reliable and easy-to-implement UQ metric.