 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnsupervised Atomic Data Mining via Multi-Kernel Graph Autoencoders for Machine Learning Force Fields
Paper and Code
Sep 15, 2025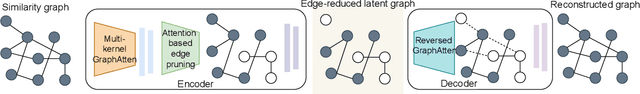
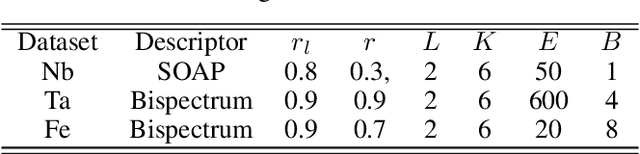
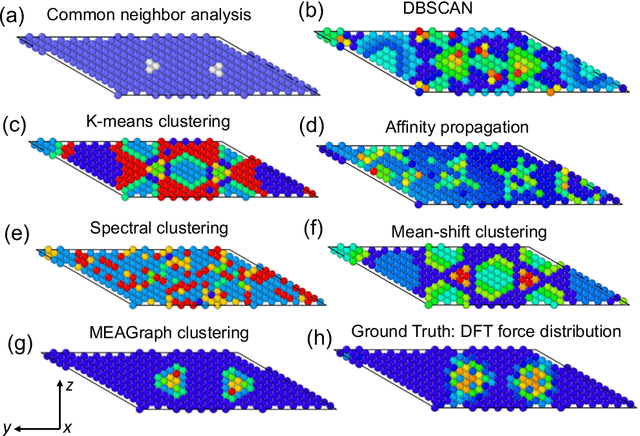
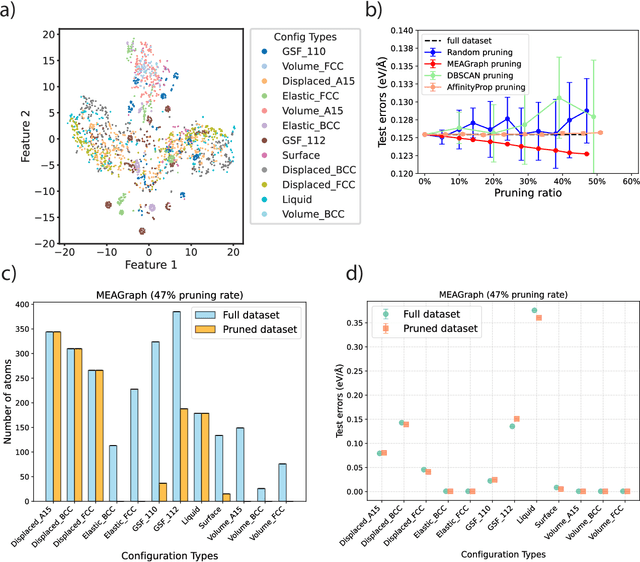
Constructing a chemically diverse dataset while avoiding sampling bias is critical to training efficient and generalizable force fields. However, in computational chemistry and materials science, many common dataset generation techniques are prone to oversampling regions of the potential energy surface. Furthermore, these regions can be difficult to identify and isolate from each other or may not align well with human intuition, making it challenging to systematically remove bias in the dataset. While traditional clustering and pruning (down-sampling) approaches can be useful for this, they can often lead to information loss or a failure to properly identify distinct regions of the potential energy surface due to difficulties associated with the high dimensionality of atomic descriptors. In this work, we introduce the Multi-kernel Edge Attention-based Graph Autoencoder (MEAGraph) model, an unsupervised approach for analyzing atomic datasets. MEAGraph combines multiple linear kernel transformations with attention-based message passing to capture geometric sensitivity and enable effective dataset pruning without relying on labels or extensive training. Demonstrated applications on niobium, tantalum, and iron datasets show that MEAGraph efficiently groups similar atomic environments, allowing for the use of basic pruning techniques for removing sampling bias. This approach provides an effective method for representation learning and clustering that can be used for data analysis, outlier detection, and dataset optimization.