 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMatbench Discovery -- An evaluation framework for machine learning crystal stability prediction
Aug 28, 2023Matbench Discovery simulates the deployment of machine learning (ML) energy models in a high-throughput search for stable inorganic crystals. We address the disconnect between (i) thermodynamic stability and formation energy and (ii) in-domain vs out-of-distribution performance. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with further insights into trade-offs between various performance metrics. To answer the question which ML methodology performs best at materials discovery, our initial release explores a variety of models including random forests, graph neural networks (GNN), one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials (UIP). Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find CHGNet > M3GNet > MACE > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi tessellation fingerprints with random forest. The top 3 models are UIPs, the winning methodology for ML-guided materials discovery, achieving F1 scores of ~0.6 for crystal stability classification and discovery acceleration factors (DAF) of up to 5x on the first 10k most stable predictions compared to dummy selection from our test set. We also highlight a sharp disconnect between commonly used global regression metrics and more task-relevant classification metrics. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV/atom above the convex hull where most materials are. Our results highlight the need to focus on classification metrics that actually correlate with improved stability hit rate.
GAUCHE: A Library for Gaussian Processes in Chemistry
Dec 06, 2022We introduce GAUCHE, a library for GAUssian processes in CHEmistry. Gaussian processes have long been a cornerstone of probabilistic machine learning, affording particular advantages for uncertainty quantification and Bayesian optimisation. Extending Gaussian processes to chemical representations, however, is nontrivial, necessitating kernels defined over structured inputs such as graphs, strings and bit vectors. By defining such kernels in GAUCHE, we seek to open the door to powerful tools for uncertainty quantification and Bayesian optimisation in chemistry. Motivated by scenarios frequently encountered in experimental chemistry, we showcase applications for GAUCHE in molecular discovery and chemical reaction optimisation. The codebase is made available at https://github.com/leojklarner/gauche
Dataset Bias in the Natural Sciences: A Case Study in Chemical Reaction Prediction and Synthesis Design
May 06, 2021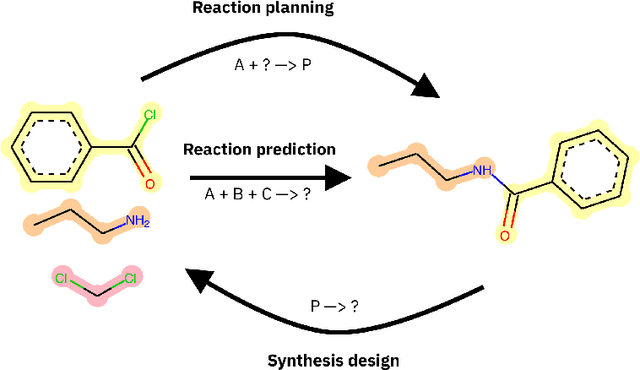
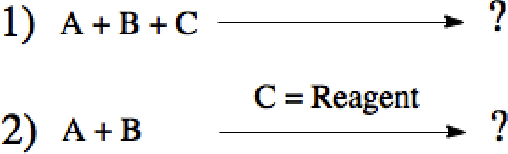
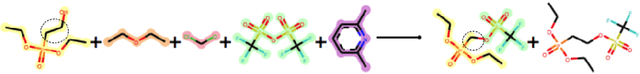
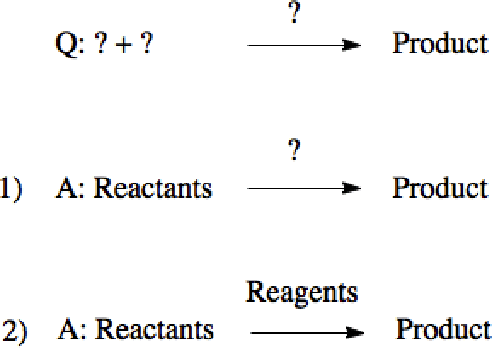
Datasets in the Natural Sciences are often curated with the goal of aiding scientific understanding and hence may not always be in a form that facilitates the application of machine learning. In this paper, we identify three trends within the fields of chemical reaction prediction and synthesis design that require a change in direction. First, the manner in which reaction datasets are split into reactants and reagents encourages testing models in an unrealistically generous manner. Second, we highlight the prevalence of mislabelled data, and suggest that the focus should be on outlier removal rather than data fitting only. Lastly, we discuss the problem of reagent prediction, in addition to reactant prediction, in order to solve the full synthesis design problem, highlighting the mismatch between what machine learning solves and what a lab chemist would need. Our critiques are also relevant to the burgeoning field of using machine learning to accelerate progress in experimental Natural Sciences, where datasets are often split in a biased way, are highly noisy, and contextual variables that are not evident from the data strongly influence the outcome of experiments.
Bayesian unsupervised learning reveals hidden structure in concentrated electrolytes
Dec 19, 2020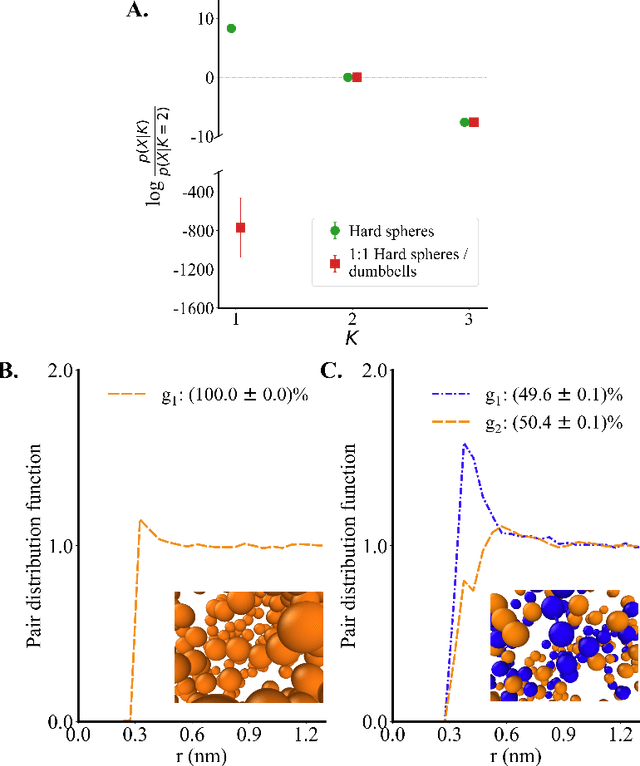
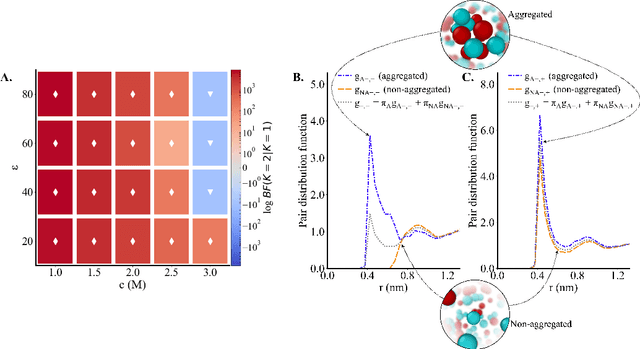
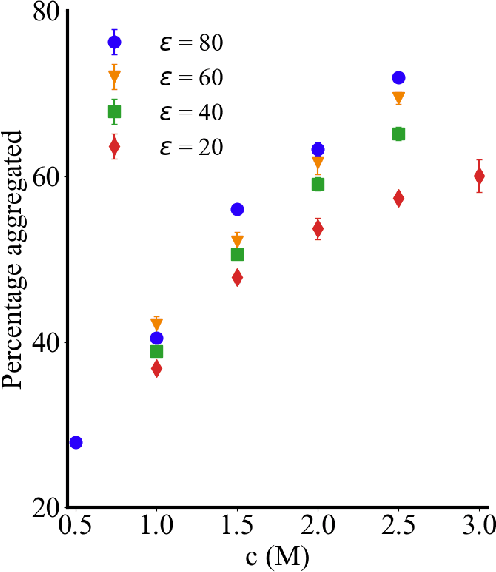
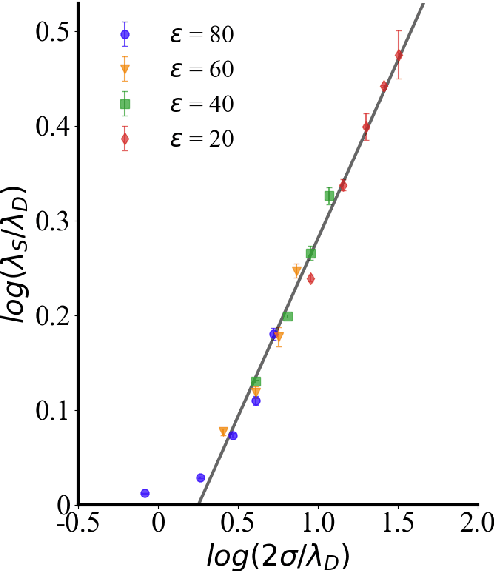
Electrolytes play an important role in a plethora of applications ranging from energy storage to biomaterials. Notwithstanding this, the structure of concentrated electrolytes remains enigmatic. Many theoretical approaches attempt to model the concentrated electrolytes by introducing the idea of ion pairs, with ions either being tightly `paired' with a counter-ion, or `free' to screen charge. In this study we reframe the problem into the language of computational statistics, and test the null hypothesis that all ions share the same local environment. Applying the framework to molecular dynamics simulations, we show that this null hypothesis is not supported by data. Our statistical technique suggests the presence of distinct local ionic environments; surprisingly, these differences arise in like charge correlations rather than unlike charge attraction. The resulting fraction of particles in non-aggregated environments shows a universal scaling behaviour across different background dielectric constants and ionic concentrations.
Investigating 3D Atomic Environments for Enhanced QSAR
Oct 24, 2020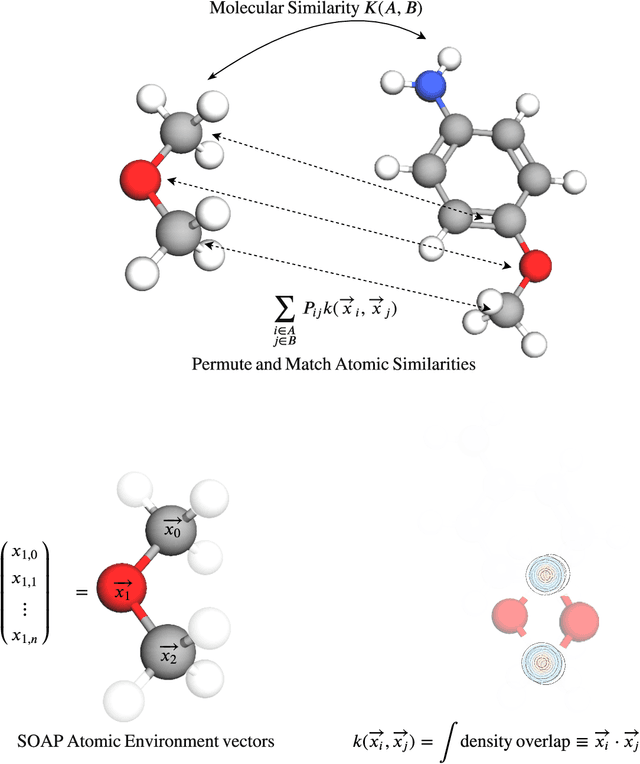
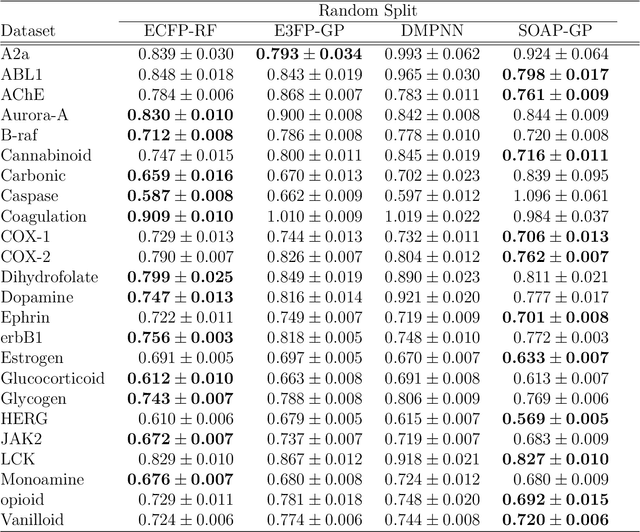
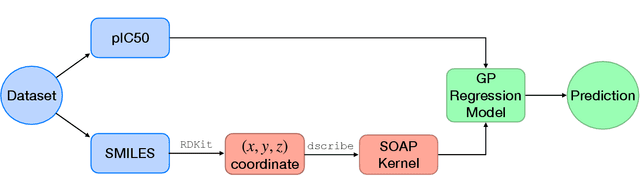
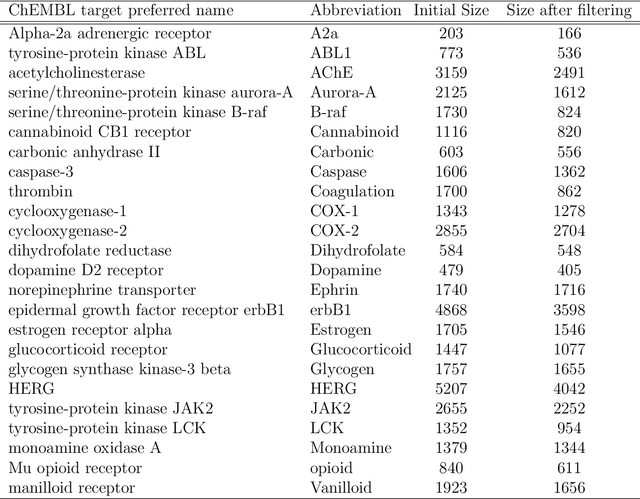
Predicting bioactivity and physical properties of molecules is a longstanding challenge in drug design. Most approaches use molecular descriptors based on a 2D representation of molecules as a graph of atoms and bonds, abstracting away the molecular shape. A difficulty in accounting for 3D shape is in designing molecular descriptors can precisely capture molecular shape while remaining invariant to rotations/translations. We describe a novel alignment-free 3D QSAR method using Smooth Overlap of Atomic Positions (SOAP), a well-established formalism developed for interpolating potential energy surfaces. We show that this approach rigorously describes local 3D atomic environments to compare molecular shapes in a principled manner. This method performs competitively with traditional fingerprint-based approaches as well as state-of-the-art graph neural networks on pIC$_{50}$ ligand-binding prediction in both random and scaffold split scenarios. We illustrate the utility of SOAP descriptors by showing that its inclusion in ensembling diverse representations statistically improves performance, demonstrating that incorporating 3D atomic environments could lead to enhanced QSAR for cheminformatics.
Predicting materials properties without crystal structure: Deep representation learning from stoichiometry
Oct 29, 2019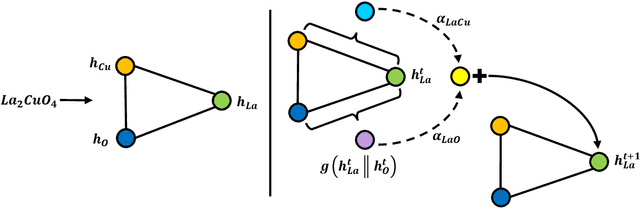
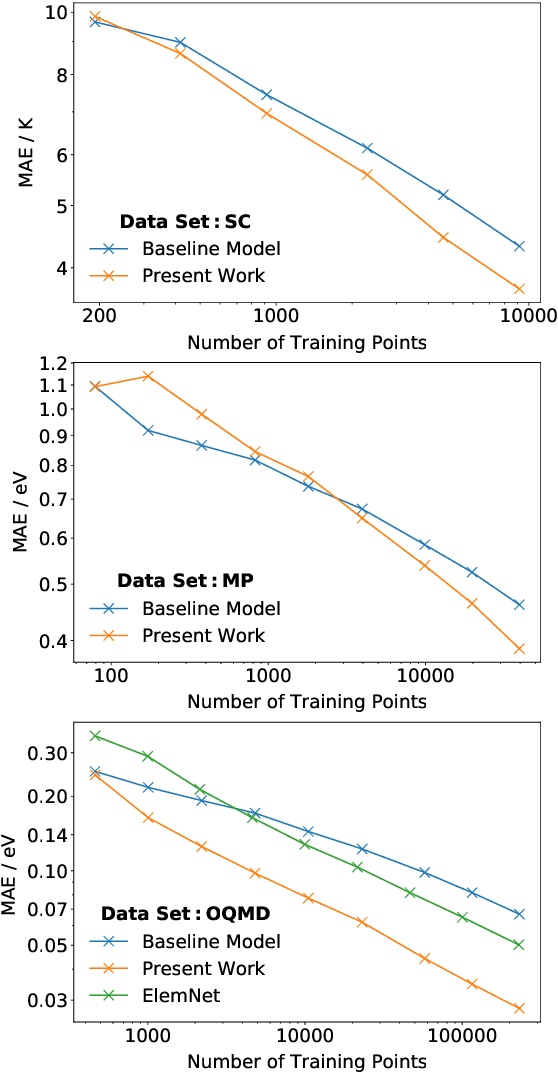
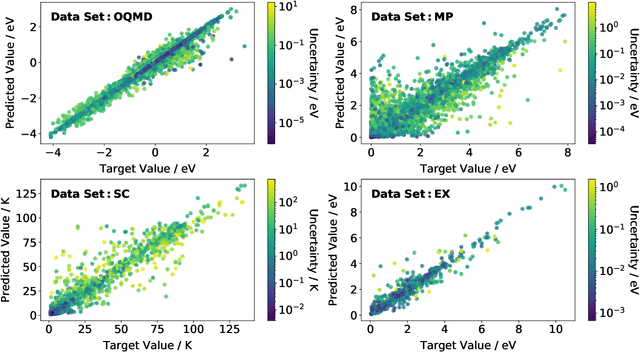
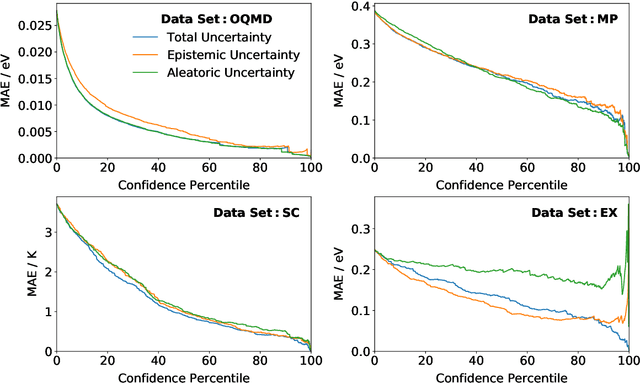
Machine learning can accelerate materials discovery by accurately predicting materials properties with low computational cost. However, the model inputs remain a key stumbling block: current methods typically use hand-engineered descriptors constructed from knowledge of either the full crystal structure -- applicable only to materials with experimentally measured structures as crystal structure prediction is computationally expensive -- or the stoichiometry. We develop a machine learning approach that takes only the stoichiometry as input and automatically learns appropriate and systematically improvable descriptors from data. Our key insight is to treat the stoichiometric formula as a dense weighted graph between elements. Compared to the state of the art, our approach achieves lower error on a plethora of challenging material properties. Moreover, our model can estimate its own uncertainty as well as transfer its learnt representation, extracting useful information from a cognate data-abundant task to deploy on a data-poor task.
Achieving Robustness to Aleatoric Uncertainty with Heteroscedastic Bayesian Optimisation
Oct 17, 2019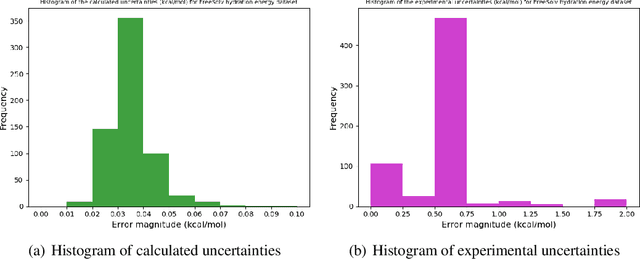
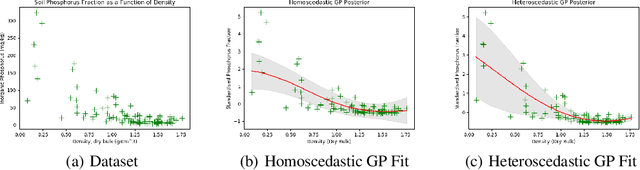
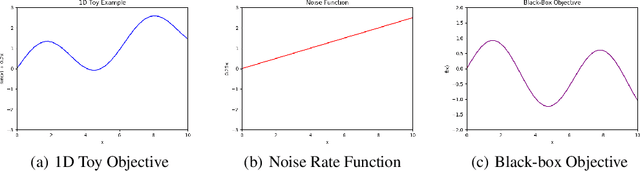
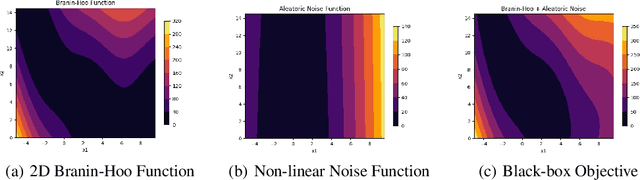
Bayesian optimisation is an important decision-making tool for high-stakes applications in drug discovery and materials design. An oft-overlooked modelling consideration however is the representation of input-dependent or heteroscedastic aleatoric uncertainty. The cost of misrepresenting this uncertainty as being homoscedastic could be high in drug discovery applications where neglecting heteroscedasticity in high throughput virtual screening could lead to a failed drug discovery program. In this paper, we propose a heteroscedastic Bayesian optimisation scheme which both represents and penalises aleatoric noise in the suggestions.Our scheme features a heteroscedastic Gaussian Process (GP) as the surrogate model in conjunction with two acquisition heuristics. First, we extend the augmented expected improvement (AEI) heuristic to the heteroscedastic setting and second, we introduce a new acquisition function, aleatoric-penalised expected improvement (ANPEI) based on a simple scalarisation of the performance and noise objective. Both methods penalise aleatoric noise in the suggestions and yield improved performance relative to a naive implementation of homoscedastic Bayesian optimisation on toy problems as well as a real-world optimisation problem.
Validating the Validation: Reanalyzing a large-scale comparison of Deep Learning and Machine Learning models for bioactivity prediction
Jun 09, 2019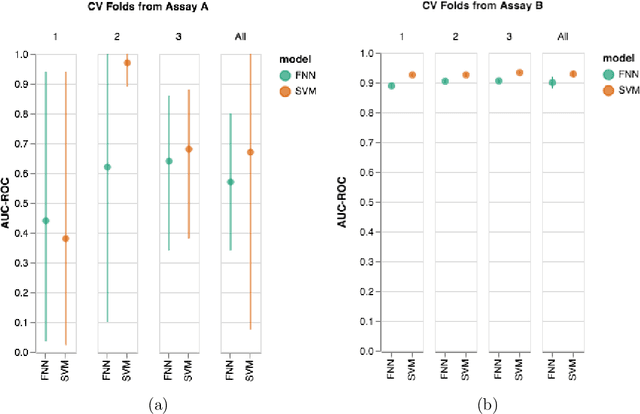
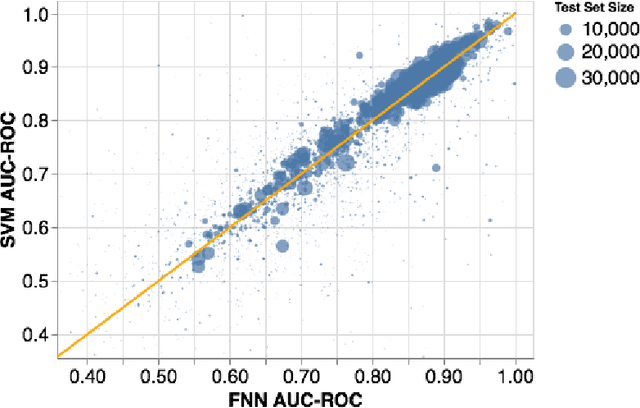
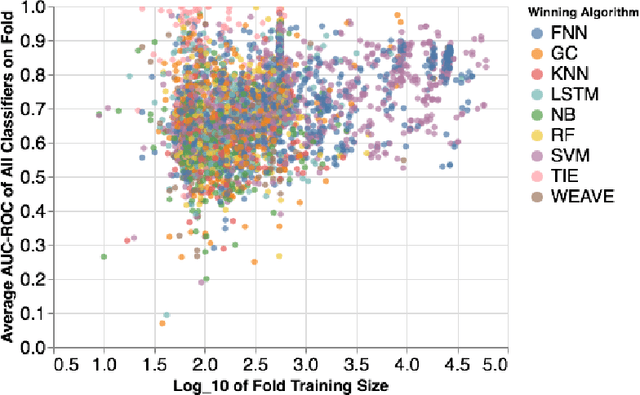
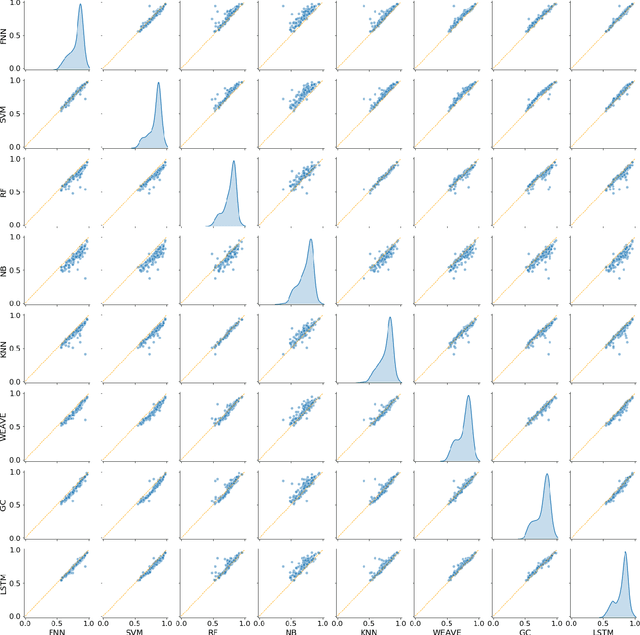
Machine learning methods may have the potential to significantly accelerate drug discovery. However, the increasing rate of new methodological approaches being published in the literature raises the fundamental question of how models should be benchmarked and validated. We reanalyze the data generated by a recently published large-scale comparison of machine learning models for bioactivity prediction and arrive at a somewhat different conclusion. We show that the performance of support vector machines is competitive with that of deep learning methods. Additionally, using a series of numerical experiments, we question the relevance of area under the receiver operating characteristic curve as a metric in virtual screening, and instead suggest that area under the precision-recall curve should be used in conjunction with the receiver operating characteristic. Our numerical experiments also highlight challenges in estimating the uncertainty in model performance via scaffold-split nested cross validation.
Bayesian semi-supervised learning for uncertainty-calibrated prediction of molecular properties and active learning
Feb 03, 2019



Predicting bioactivity and physical properties of small molecules is a central challenge in drug discovery. Deep learning is becoming the method of choice but studies to date focus on mean accuracy as the main metric. However, to replace costly and mission-critical experiments by models, a high mean accuracy is not enough: Outliers can derail a discovery campaign, thus models need reliably predict when it will fail, even when the training data is biased; experiments are expensive, thus models need to be data-efficient and suggest informative training sets using active learning. We show that uncertainty quantification and active learning can be achieved by Bayesian semi-supervised graph convolutional neural networks. The Bayesian approach estimates uncertainty in a statistically principled way through sampling from the posterior distribution. Semi-supervised learning disentangles representation learning and regression, keeping uncertainty estimates accurate in the low data limit and allowing the model to start active learning from a small initial pool of training data. Our study highlights the promise of Bayesian deep learning for chemistry.
Geometry of energy landscapes and the optimizability of deep neural networks
Aug 01, 2018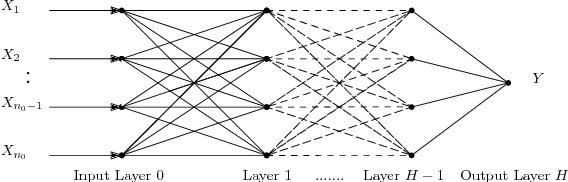
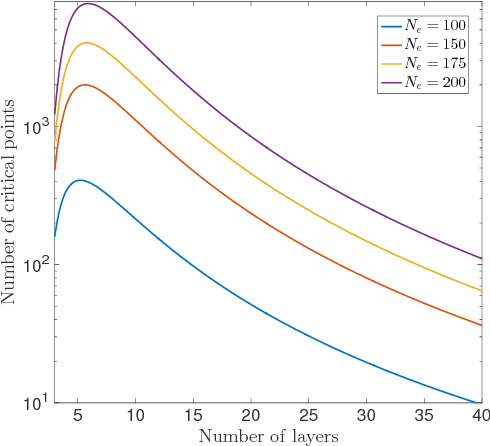
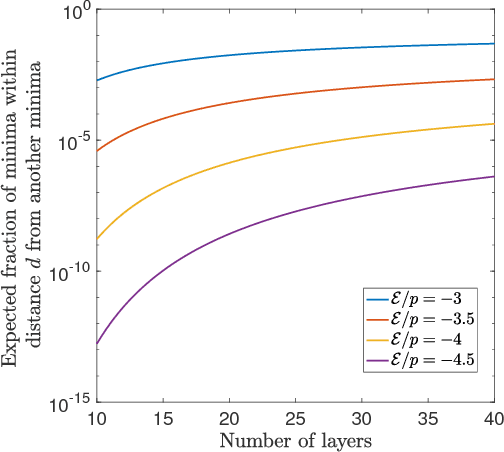
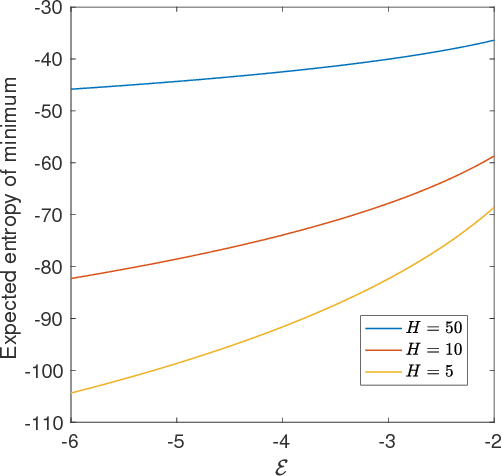
Deep neural networks are workhorse models in machine learning with multiple layers of non-linear functions composed in series. Their loss function is highly non-convex, yet empirically even gradient descent minimisation is sufficient to arrive at accurate and predictive models. It is hitherto unknown why are deep neural networks easily optimizable. We analyze the energy landscape of a spin glass model of deep neural networks using random matrix theory and algebraic geometry. We analytically show that the multilayered structure holds the key to optimizability: Fixing the number of parameters and increasing network depth, the number of stationary points in the loss function decreases, minima become more clustered in parameter space, and the tradeoff between the depth and width of minima becomes less severe. Our analytical results are numerically verified through comparison with neural networks trained on a set of classical benchmark datasets. Our model uncovers generic design principles of machine learning models.