 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWavelet regression and additive models for irregularly spaced data
Mar 11, 2019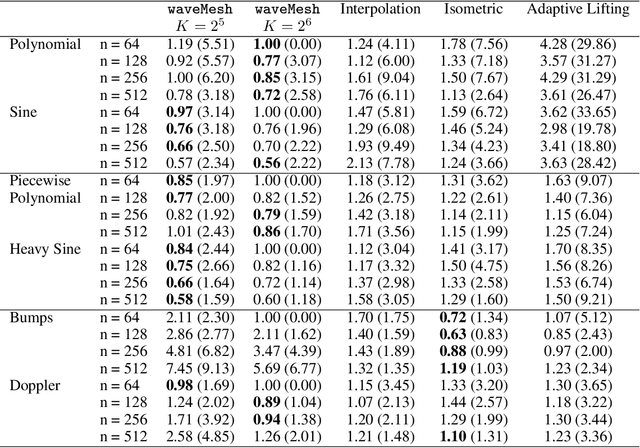
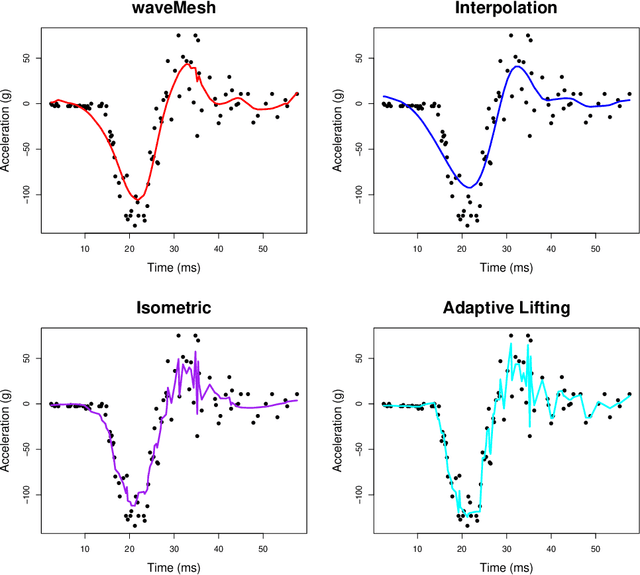

We present a novel approach for nonparametric regression using wavelet basis functions. Our proposal, $\texttt{waveMesh}$, can be applied to non-equispaced data with sample size not necessarily a power of 2. We develop an efficient proximal gradient descent algorithm for computing the estimator and establish adaptive minimax convergence rates. The main appeal of our approach is that it naturally extends to additive and sparse additive models for a potentially large number of covariates. We prove minimax optimal convergence rates under a weak compatibility condition for sparse additive models. The compatibility condition holds when we have a small number of covariates. Additionally, we establish convergence rates for when the condition is not met. We complement our theoretical results with empirical studies comparing $\texttt{waveMesh}$ to existing methods.
Generalized Score Matching for Non-Negative Data
Dec 26, 2018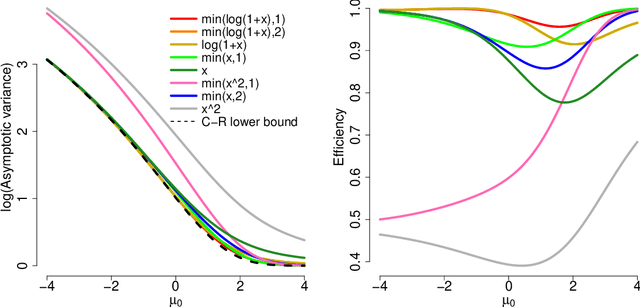
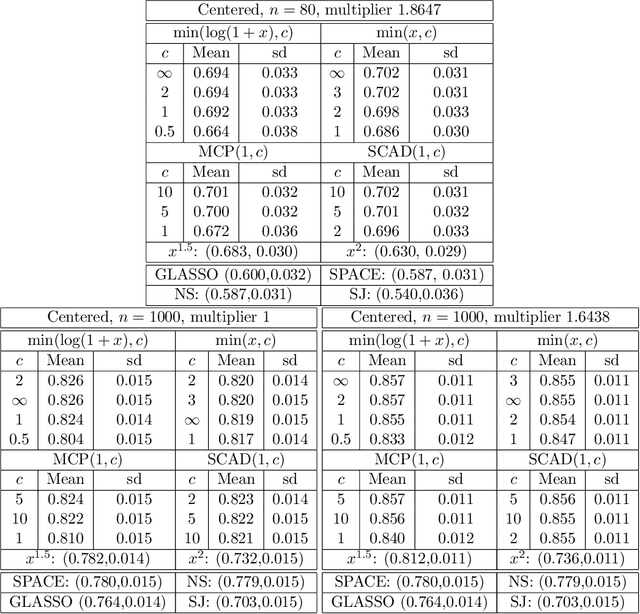
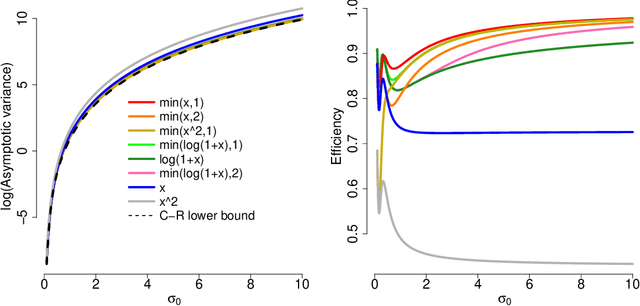
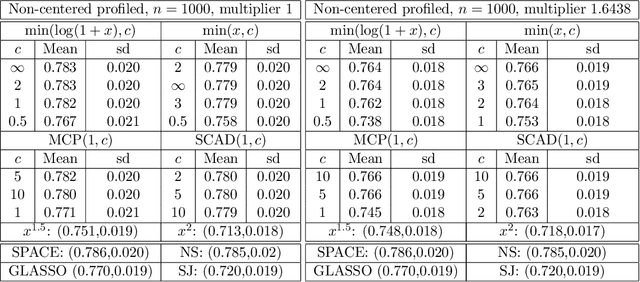
A common challenge in estimating parameters of probability density functions is the intractability of the normalizing constant. While in such cases maximum likelihood estimation may be implemented using numerical integration, the approach becomes computationally intensive. The score matching method of Hyv\"arinen [2005] avoids direct calculation of the normalizing constant and yields closed-form estimates for exponential families of continuous distributions over $\mathbb{R}^m$. Hyv\"arinen [2007] extended the approach to distributions supported on the non-negative orthant, $\mathbb{R}_+^m$. In this paper, we give a generalized form of score matching for non-negative data that improves estimation efficiency. As an example, we consider a general class of pairwise interaction models. Addressing an overlooked inexistence problem, we generalize the regularized score matching method of Lin et al. [2016] and improve its theoretical guarantees for non-negative Gaussian graphical models.
An Interpretable and Sparse Neural Network Model for Nonlinear Granger Causality Discovery
Jun 25, 2018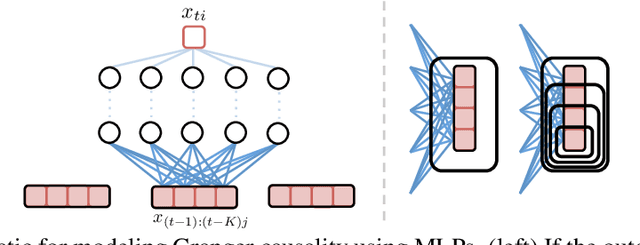
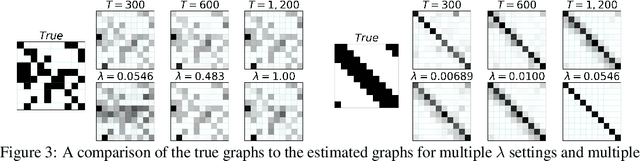
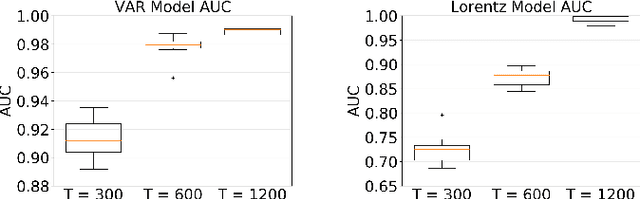
While most classical approaches to Granger causality detection repose upon linear time series assumptions, many interactions in neuroscience and economics applications are nonlinear. We develop an approach to nonlinear Granger causality detection using multilayer perceptrons where the input to the network is the past time lags of all series and the output is the future value of a single series. A sufficient condition for Granger non-causality in this setting is that all of the outgoing weights of the input data, the past lags of a series, to the first hidden layer are zero. For estimation, we utilize a group lasso penalty to shrink groups of input weights to zero. We also propose a hierarchical penalty for simultaneous Granger causality and lag estimation. We validate our approach on simulated data from both a sparse linear autoregressive model and the sparse and nonlinear Lorenz-96 model.
An Efficient ADMM Algorithm for Structural Break Detection in Multivariate Time Series
Jun 25, 2018
We present an efficient alternating direction method of multipliers (ADMM) algorithm for segmenting a multivariate non-stationary time series with structural breaks into stationary regions. We draw from recent work where the series is assumed to follow a vector autoregressive model within segments and a convex estimation procedure may be formulated using group fused lasso penalties. Our ADMM approach first splits the convex problem into a global quadratic program and a simple group lasso proximal update. We show that the global problem may be parallelized over rows of the time dependent transition matrices and furthermore that each subproblem may be rewritten in a form identical to the log-likelihood of a Gaussian state space model. Consequently, we develop a Kalman smoothing algorithm to solve the global update in time linear in the length of the series.
The Reduced PC-Algorithm: Improved Causal Structure Learning in Large Random Networks
Jun 16, 2018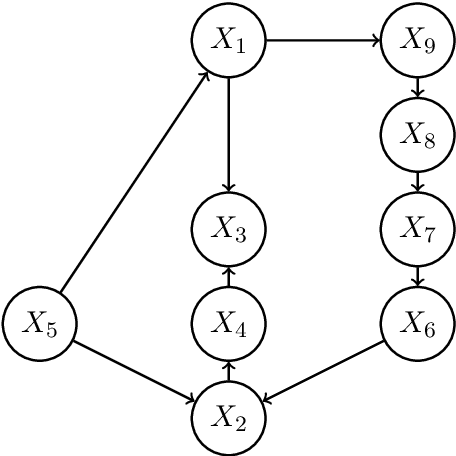
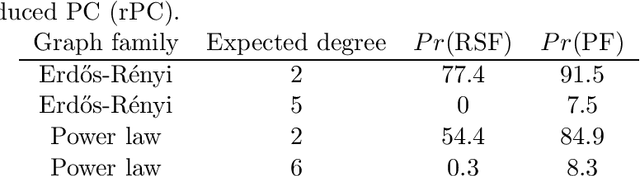
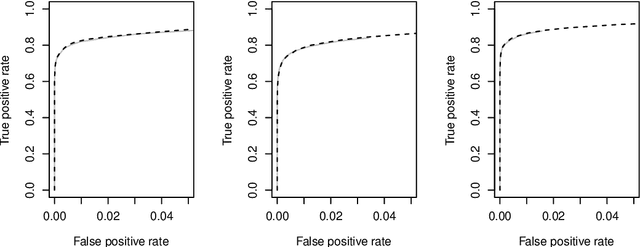

We consider the task of estimating a high-dimensional directed acyclic graph, given observations from a linear structural equation model with arbitrary noise distribution. By exploiting properties of common random graphs, we develop a new algorithm that requires conditioning only on small sets of variables. The proposed algorithm, which is essentially a modified version of the PC-Algorithm, offers significant gains in both computational complexity and estimation accuracy. In particular, it results in more efficient and accurate estimation in large networks containing hub nodes, which are common in biological systems. We prove the consistency of the proposed algorithm, and show that it also requires a less stringent faithfulness assumption than the PC-Algorithm. Simulations in low and high-dimensional settings are used to illustrate these findings. An application to gene expression data suggests that the proposed algorithm can identify a greater number of clinically relevant genes than current methods.
Neural Granger Causality for Nonlinear Time Series
Feb 16, 2018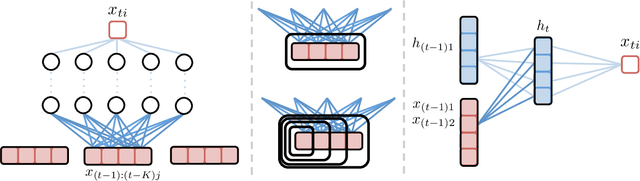
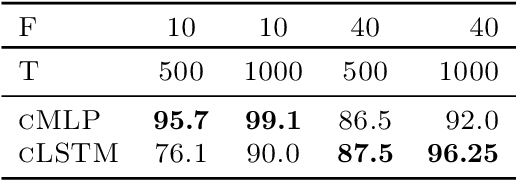
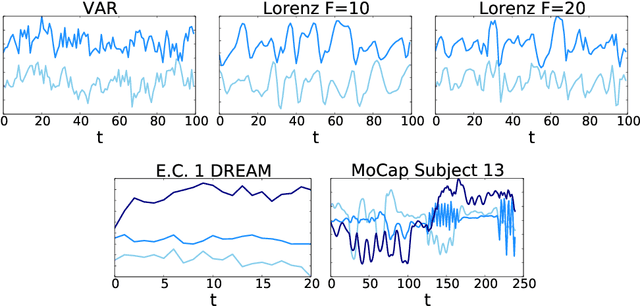

While most classical approaches to Granger causality detection assume linear dynamics, many interactions in applied domains, like neuroscience and genomics, are inherently nonlinear. In these cases, using linear models may lead to inconsistent estimation of Granger causal interactions. We propose a class of nonlinear methods by applying structured multilayer perceptrons (MLPs) or recurrent neural networks (RNNs) combined with sparsity-inducing penalties on the weights. By encouraging specific sets of weights to be zero---in particular through the use of convex group-lasso penalties---we can extract the Granger causal structure. To further contrast with traditional approaches, our framework naturally enables us to efficiently capture long-range dependencies between series either via our RNNs or through an automatic lag selection in the MLP. We show that our neural Granger causality methods outperform state-of-the-art nonlinear Granger causality methods on the DREAM3 challenge data. This data consists of nonlinear gene expression and regulation time courses with only a limited number of time points. The successes we show in this challenging dataset provide a powerful example of how deep learning can be useful in cases that go beyond prediction on large datasets. We likewise demonstrate our methods in detecting nonlinear interactions in a human motion capture dataset.
In Defense of the Indefensible: A Very Naive Approach to High-Dimensional Inference
Dec 07, 2017
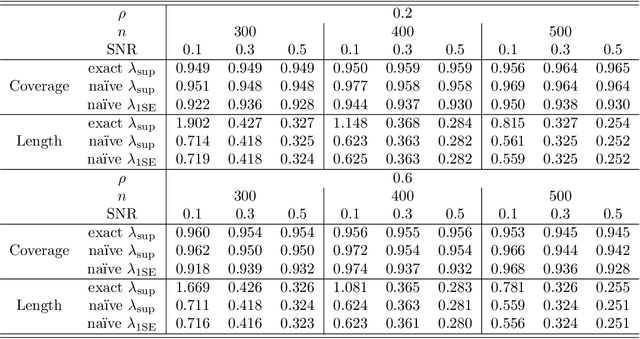
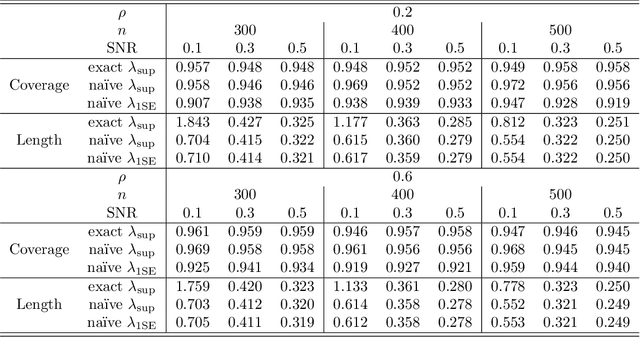
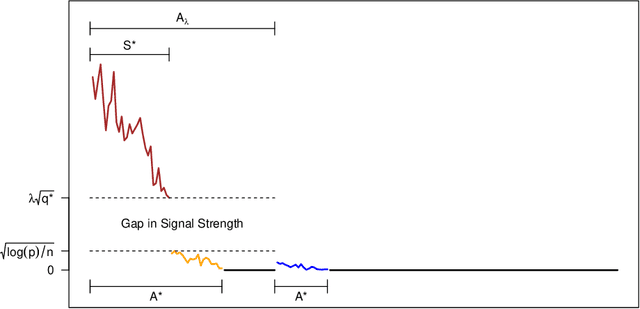
In recent years, a great deal of interest has focused on conducting inference on the parameters in a linear model in the high-dimensional setting. In this paper, we consider a simple and very na\"{i}ve two-step procedure for this task, in which we (i) fit a lasso model in order to obtain a subset of the variables; and (ii) fit a least squares model on the lasso-selected set. Conventional statistical wisdom tells us that we cannot make use of the standard statistical inference tools for the resulting least squares model (such as confidence intervals and $p$-values), since we peeked at the data twice: once in running the lasso, and again in fitting the least squares model. However, in this paper, we show that under a certain set of assumptions, with high probability, the set of variables selected by the lasso is deterministic. Consequently, the na\"{i}ve two-step approach can yield confidence intervals that have asymptotically correct coverage, as well as p-values with proper Type-I error control. Furthermore, this two-step approach unifies two existing camps of work on high-dimensional inference: one camp has focused on inference based on a sub-model selected by the lasso, and the other has focused on inference using a debiased version of the lasso estimator.
Nonparametric Regression with Adaptive Truncation via a Convex Hierarchical Penalty
Dec 01, 2016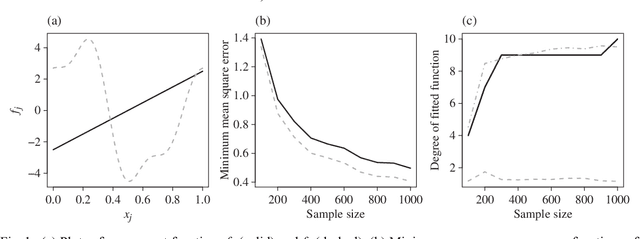
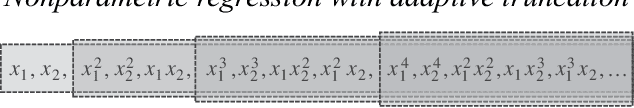
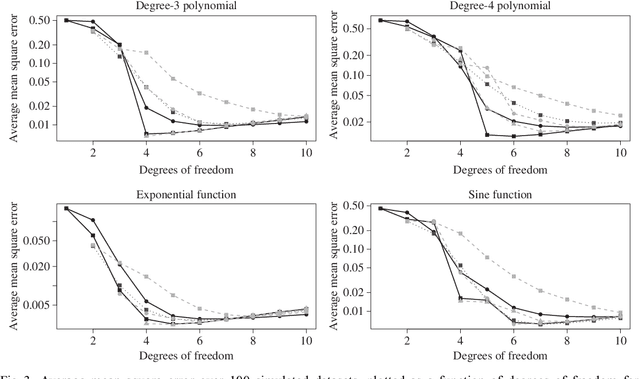
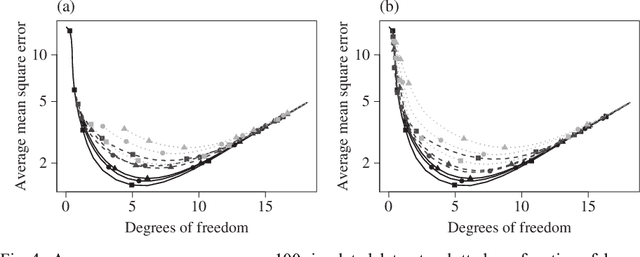
We consider the problem of non-parametric regression with a potentially large number of covariates. We propose a convex, penalized estimation framework that is particularly well-suited for high-dimensional sparse additive models. The proposed approach combines appealing features of finite basis representation and smoothing penalties for non-parametric estimation. In particular, in the case of additive models, a finite basis representation provides a parsimonious representation for fitted functions but is not adaptive when component functions posses different levels of complexity. On the other hand, a smoothing spline type penalty on the component functions is adaptive but does not offer a parsimonious representation of the estimated function. The proposed approach simultaneously achieves parsimony and adaptivity in a computationally efficient framework. We demonstrate these properties through empirical studies on both real and simulated datasets. We show that our estimator converges at the minimax rate for functions within a hierarchical class. We further establish minimax rates for a large class of sparse additive models. The proposed method is implemented using an efficient algorithm that scales similarly to the Lasso with the number of covariates and samples size.
Joint Estimation of Precision Matrices in Heterogeneous Populations
Jan 02, 2016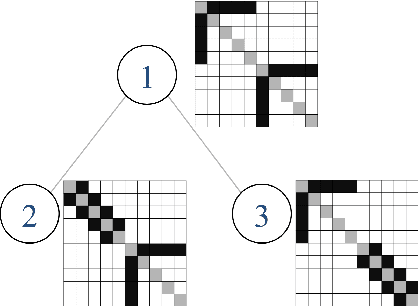
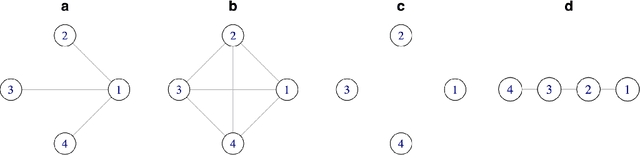
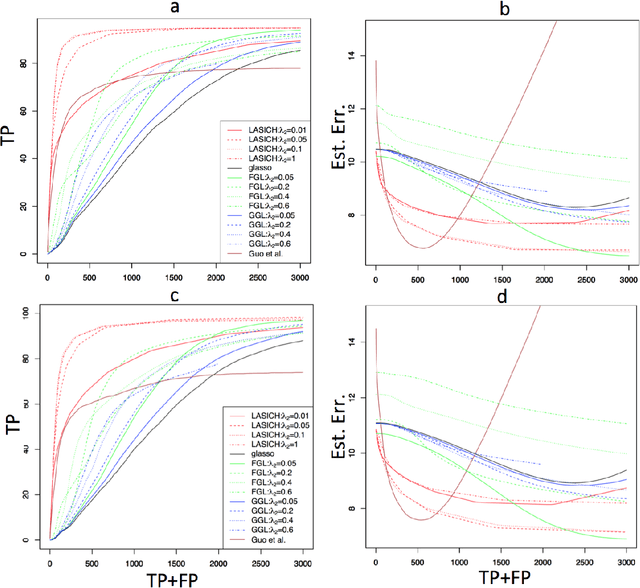
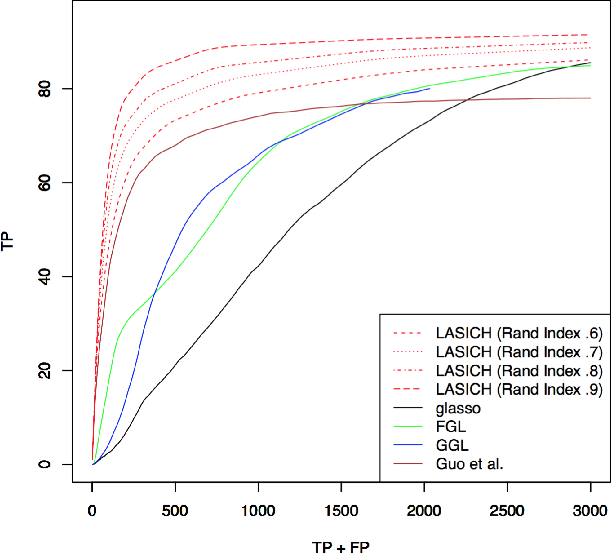
We introduce a general framework for estimation of inverse covariance, or precision, matrices from heterogeneous populations. The proposed framework uses a Laplacian shrinkage penalty to encourage similarity among estimates from disparate, but related, subpopulations, while allowing for differences among matrices. We propose an efficient alternating direction method of multipliers (ADMM) algorithm for parameter estimation, as well as its extension for faster computation in high dimensions by thresholding the empirical covariance matrix to identify the joint block diagonal structure in the estimated precision matrices. We establish both variable selection and norm consistency of the proposed estimator for distributions with exponential or polynomial tails. Further, to extend the applicability of the method to the settings with unknown populations structure, we propose a Laplacian penalty based on hierarchical clustering, and discuss conditions under which this data-driven choice results in consistent estimation of precision matrices in heterogenous populations. Extensive numerical studies and applications to gene expression data from subtypes of cancer with distinct clinical outcomes indicate the potential advantages of the proposed method over existing approaches.
Inference in High Dimensions with the Penalized Score Test
May 19, 2014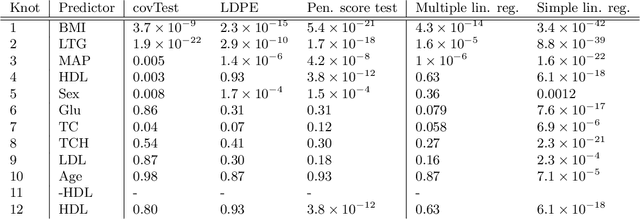
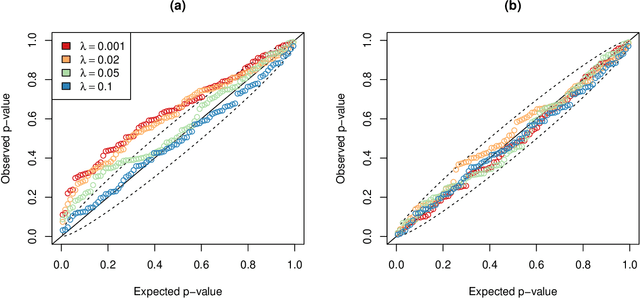
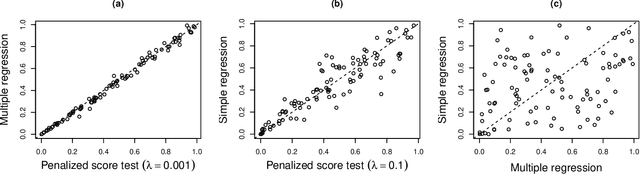
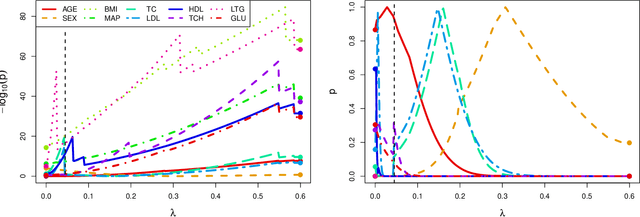
In recent years, there has been considerable theoretical development regarding variable selection consistency of penalized regression techniques, such as the lasso. However, there has been relatively little work on quantifying the uncertainty in these selection procedures. In this paper, we propose a new method for inference in high dimensions using a score test based on penalized regression. In this test, we perform penalized regression of an outcome on all but a single feature, and test for correlation of the residuals with the held-out feature. This procedure is applied to each feature in turn. Interestingly, when an $\ell_1$ penalty is used, the sparsity pattern of the lasso corresponds exactly to a decision based on the proposed test. Further, when an $\ell_2$ penalty is used, the test corresponds precisely to a score test in a mixed effects model, in which the effects of all but one feature are assumed to be random. We formulate the hypothesis being tested as a compromise between the null hypotheses tested in simple linear regression on each feature and in multiple linear regression on all features, and develop reference distributions for some well-known penalties. We also examine the behavior of the test on real and simulated data.