 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobustness of deep learning classification to adversarial input on GPUs: asynchronous parallel accumulation is a source of vulnerability
Mar 21, 2025



The ability of machine learning (ML) classification models to resist small, targeted input perturbations - known as adversarial attacks - is a key measure of their safety and reliability. We show that floating-point non associativity (FPNA) coupled with asynchronous parallel programming on GPUs is sufficient to result in misclassification, without any perturbation to the input. Additionally, we show this misclassification is particularly significant for inputs close to the decision boundary and that standard adversarial robustness results may be overestimated up to 4.6% when not considering machine-level details. We first study a linear classifier, before focusing on standard Graph Neural Network (GNN) architectures and datasets. We present a novel black-box attack using Bayesian optimization to determine external workloads that bias the output of reductions on GPUs and reliably lead to misclassification. Motivated by these results, we present a new learnable permutation (LP) gradient-based approach, to learn floating point operation orderings that lead to misclassifications, making the assumption that any reduction or permutation ordering is possible. This LP approach provides a worst-case estimate in a computationally efficient manner, avoiding the need to run identical experiments tens of thousands of times over a potentially large set of possible GPU states or architectures. Finally, we investigate parallel reduction ordering across different GPU architectures for a reduction under three conditions: (1) executing external background workloads, (2) utilizing multi-GPU virtualization, and (3) applying power capping. Our results demonstrate that parallel reduction ordering varies significantly across architectures under the first two conditions. The results and methods developed here can help to include machine-level considerations into adversarial robustness assessments.
Impacts of floating-point non-associativity on reproducibility for HPC and deep learning applications
Aug 09, 2024Run-by-run variability in parallel programs caused by floating-point non-associativity (FPNA) has been known to significantly affect reproducibility in iterative algorithms, due to accumulating errors. Non-reproducibility negatively affects efficiency and effectiveness of correctness testing for stochastic programs. Recently, the sensitivity of deep learning (DL) training and inference pipelines to FPNA have been found to be extreme, and can prevent certification for commercial applications, accurate assessment of robustness and sensitivity, and bug detection. New approaches in scientific computing applications have coupled DL models with high-performance computing (HPC) simulations, leading to an aggravation of debugging and testing challenges. Here we perform an investigation of the statistical properties of FPNA within modern parallel programming models, analyze performance and productivity impacts of replacing atomic operations with deterministic alternatives on GPUs, and examine the recently-added deterministic options within the PyTorch framework within the context of GPU deployment, uncovering and quantifying the impacts of input parameters triggering run-by-run variability and reporting on the reliability and completeness of the documentation. Finally, we evaluate the strategy of exploiting automatic determinism provided by deterministic hardware, using the Groq LPU$^{TM}$ accelerator for inference portions of the DL pipeline. We demonstrate the benefits that this strategy can provide within reproducibility and correctness efforts.
DIPS-Plus: The Enhanced Database of Interacting Protein Structures for Interface Prediction
Jun 06, 2021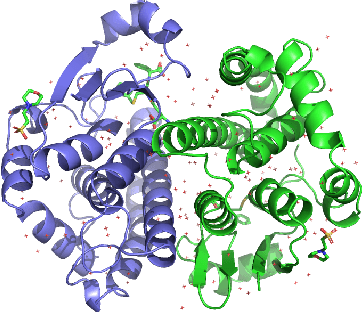

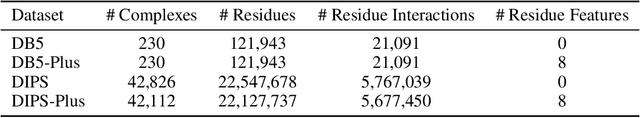

How and where proteins interface with one another can ultimately impact the proteins' functions along with a range of other biological processes. As such, precise computational methods for protein interface prediction (PIP) come highly sought after as they could yield significant advances in drug discovery and design as well as protein function analysis. However, the traditional benchmark dataset for this task, Docking Benchmark 5 (DB5), contains only a paltry 230 complexes for training, validating, and testing different machine learning algorithms. In this work, we expand on a dataset recently introduced for this task, the Database of Interacting Protein Structures (DIPS), to present DIPS-Plus, an enhanced, feature-rich dataset of 42,112 complexes for geometric deep learning of protein interfaces. The previous version of DIPS contains only the Cartesian coordinates and types of the atoms comprising a given protein complex, whereas DIPS-Plus now includes a plethora of new residue-level features including protrusion indices, half-sphere amino acid compositions, and new profile hidden Markov model (HMM)-based sequence features for each amino acid, giving researchers a large, well-curated feature bank for training protein interface prediction methods.