 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Large-Time-Step Molecular Dynamics with Graph Neural Networks
Paper and Code
Dec 21, 2021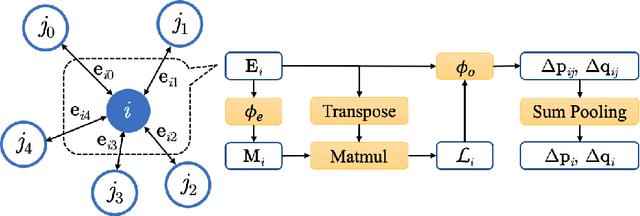

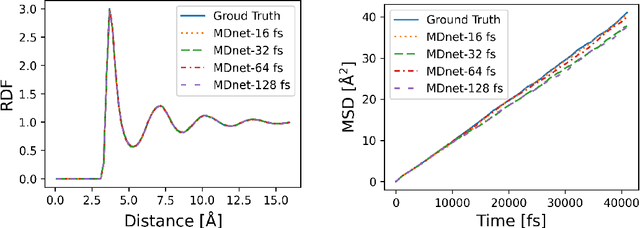
Molecular dynamics (MD) simulation predicts the trajectory of atoms by solving Newton's equation of motion with a numeric integrator. Due to physical constraints, the time step of the integrator need to be small to maintain sufficient precision. This limits the efficiency of simulation. To this end, we introduce a graph neural network (GNN) based model, MDNet, to predict the evolution of coordinates and momentum with large time steps. In addition, MDNet can easily scale to a larger system, due to its linear complexity with respect to the system size. We demonstrate the performance of MDNet on a 4000-atom system with large time steps, and show that MDNet can predict good equilibrium and transport properties, well aligned with standard MD simulations.