 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransfer Learning for Protein Structure Classification at Low Resolution
Paper and Code
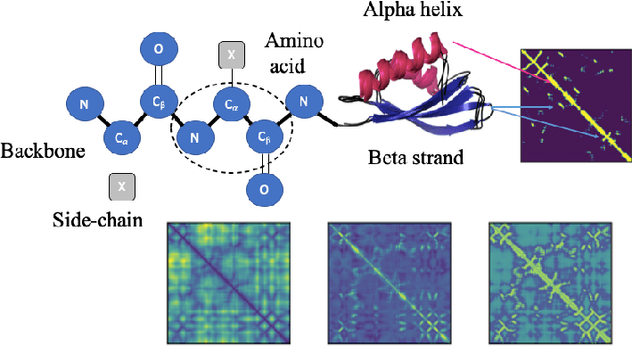
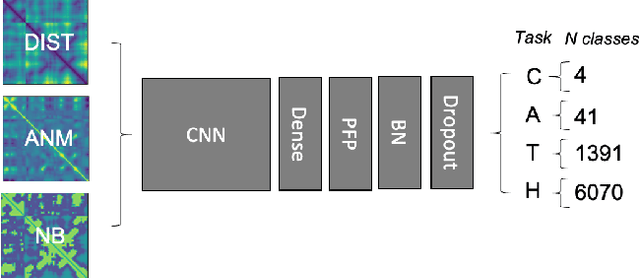
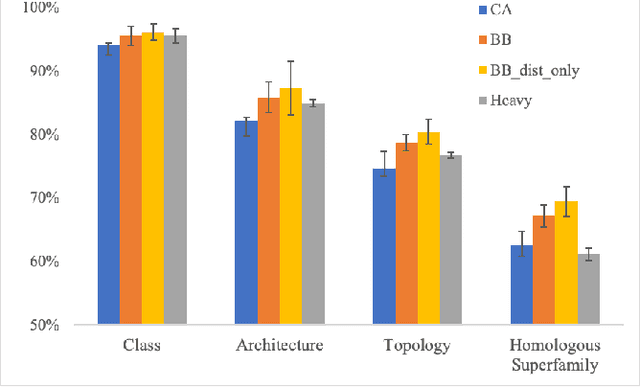

Structure determination is key to understanding protein function at a molecular level. Whilst significant advances have been made in predicting structure and function from amino acid sequence, researchers must still rely on expensive, time-consuming analytical methods to visualise detailed protein conformation. In this study, we demonstrate that it is possible to make accurate ($\geq$80%) predictions of protein class and architecture from structures determined at low ($>$3A) resolution, using a deep convolutional neural network trained on high-resolution ($\leq$3A) structures represented as 2D matrices. Thus, we provide proof of concept for high-speed, low-cost protein structure classification at low resolution, and a basis for extension to prediction of function. We investigate the impact of the input representation on classification performance, showing that side-chain information may not be necessary for fine-grained structure predictions. Finally, we confirm that high-resolution, low-resolution and NMR-determined structures inhabit a common feature space, and thus provide a theoretical foundation for boosting with single-image super-resolution.