 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModerate Adaptive Linear Units (MoLU)
Feb 28, 2023



We propose a new high-performance activation function, Moderate Adaptive Linear Units (MoLU), for the deep neural network. The MoLU is a simple, beautiful and powerful activation function that can be a good main activation function among hundreds of activation functions. Because the MoLU is made up of the elementary functions, not only it is a infinite diffeomorphism (i.e. smooth and infinitely differentiable over whole domains), but also it decreases training time.
Homotopy-based training of NeuralODEs for accurate dynamics discovery
Oct 04, 2022



Conceptually, Neural Ordinary Differential Equations (NeuralODEs) pose an attractive way to extract dynamical laws from time series data, as they are natural extensions of the traditional differential equation-based modeling paradigm of the physical sciences. In practice, NeuralODEs display long training times and suboptimal results, especially for longer duration data where they may fail to fit the data altogether. While methods have been proposed to stabilize NeuralODE training, many of these involve placing a strong constraint on the functional form the trained NeuralODE can take that the actual underlying governing equation does not guarantee satisfaction. In this work, we present a novel NeuralODE training algorithm that leverages tools from the chaos and mathematical optimization communities - synchronization and homotopy optimization - for a breakthrough in tackling the NeuralODE training obstacle. We demonstrate architectural changes are unnecessary for effective NeuralODE training. Compared to the conventional training methods, our algorithm achieves drastically lower loss values without any changes to the model architectures. Experiments on both simulated and real systems with complex temporal behaviors demonstrate NeuralODEs trained with our algorithm are able to accurately capture true long term behaviors and correctly extrapolate into the future.
Bayesian neural network with pretrained protein embedding enhances prediction accuracy of drug-protein interaction
Dec 21, 2020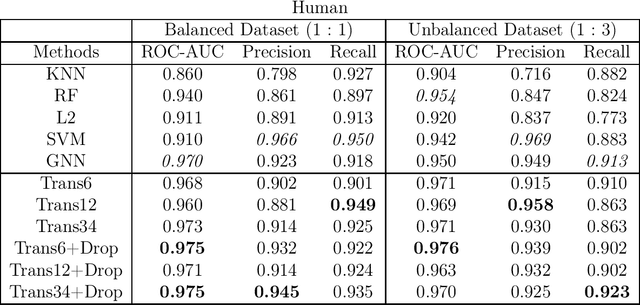
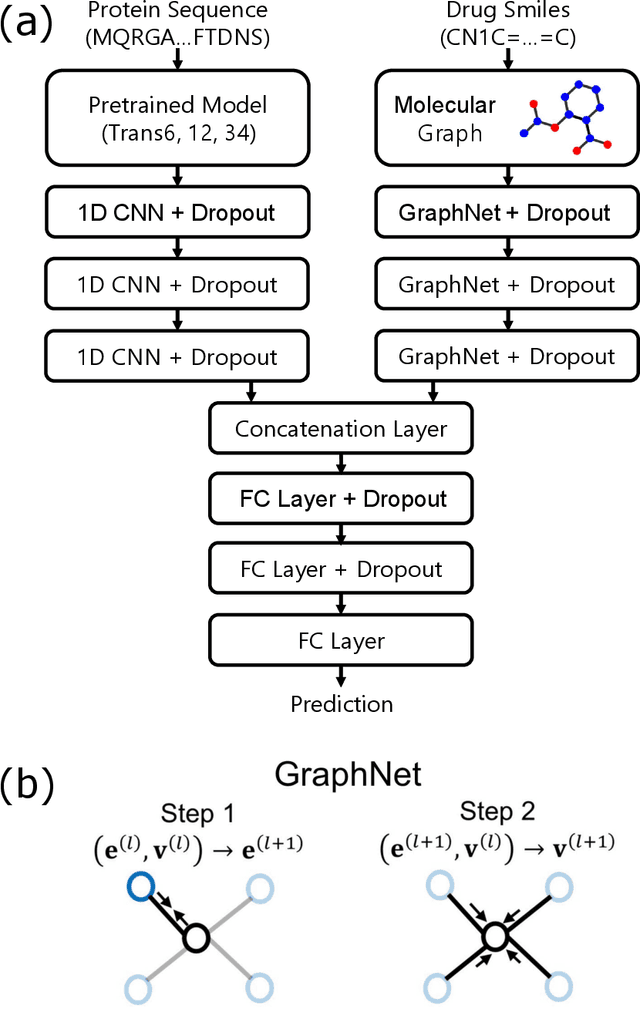
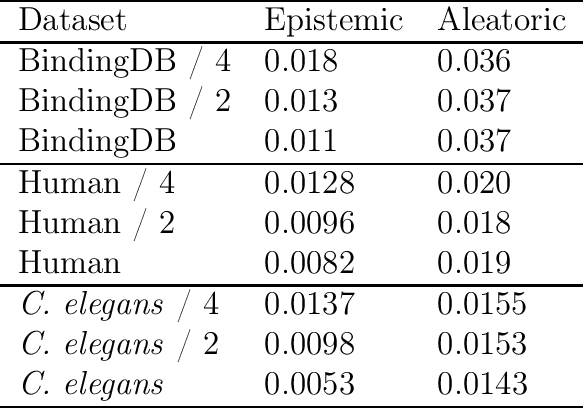
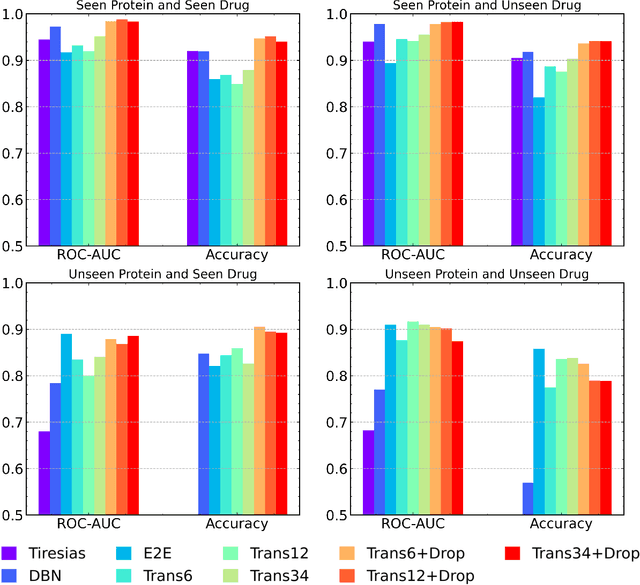
The characterization of drug-protein interactions is crucial in the high-throughput screening for drug discovery. The deep learning-based approaches have attracted attention because they can predict drug-protein interactions without trial-and-error by humans. However, because data labeling requires significant resources, the available protein data size is relatively small, which consequently decreases model performance. Here we propose two methods to construct a deep learning framework that exhibits superior performance with a small labeled dataset. At first, we use transfer learning in encoding protein sequences with a pretrained model, which trains general sequence representations in an unsupervised manner. Second, we use a Bayesian neural network to make a robust model by estimating the data uncertainty. As a result, our model performs better than the previous baselines for predicting drug-protein interactions. We also show that the quantified uncertainty from the Bayesian inference is related to the confidence and can be used for screening DPI data points.