 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Knowledge to Action: Outcomes of the 2025 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
May 04, 2026Large language models (LLMs) are rapidly changing how researchers in materials science and chemistry discover, organize, and act on scientific knowledge. This paper analyzes a broad set of community-developed LLM applications in an effort to identify emerging patterns in how these systems can be used across the scientific research lifecycle. We organize the projects into two complementary categories: Knowledge Infrastructure, systems that structure, retrieve, synthesize, and validate scientific information; and Action Systems, systems that execute, coordinate, or automate scientific work across computational and experimental environments. The submissions reveal a shift from single-purpose LLM tools toward integrated, multi-agent workflows that combine retrieval, reasoning, tool use, and domain-specific validation. Prominent themes include retrieval-augmented generation as grounding infrastructure, persistent structured knowledge representations, multimodal and multilingual scientific inputs, and early progress toward laboratory-integrated closed-loop systems. Together, these results suggest that LLMs are evolving from general-purpose assistants into composable infrastructure for scientific reasoning and action. This work provides a community snapshot of that transition and a practical taxonomy for understanding emerging LLM-enabled workflows in materials science and chemistry.
Analyzing dynamical disorder for charge transport in organic semiconductors via machine learning
Feb 02, 2021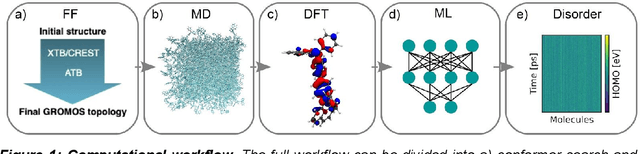
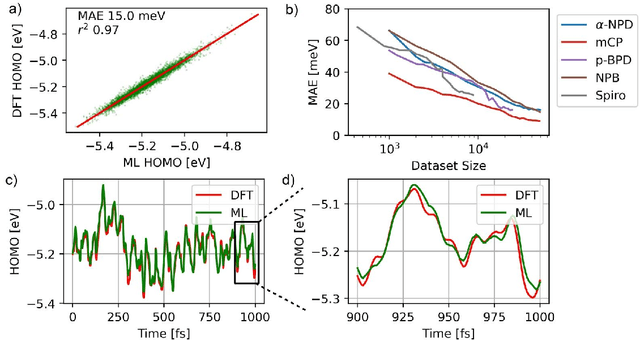
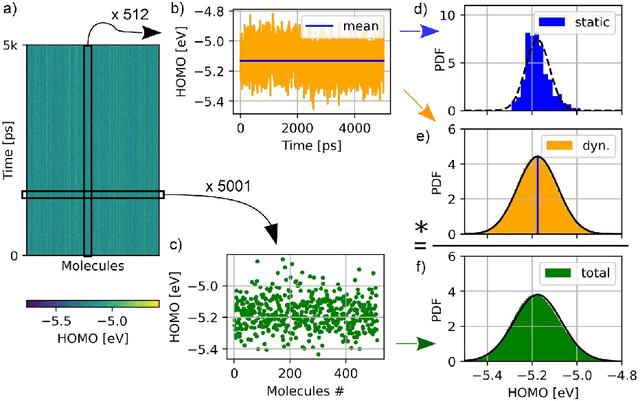
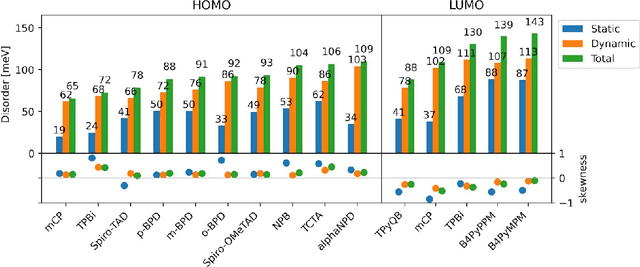
Organic semiconductors are indispensable for today's display technologies in form of organic light emitting diodes (OLEDs) and further optoelectronic applications. However, organic materials do not reach the same charge carrier mobility as inorganic semiconductors, limiting the efficiency of devices. To find or even design new organic semiconductors with higher charge carrier mobility, computational approaches, in particular multiscale models, are becoming increasingly important. However, such models are computationally very costly, especially when large systems and long time scales are required, which is the case to compute static and dynamic energy disorder, i.e. dominant factor to determine charge transport. Here we overcome this drawback by integrating machine learning models into multiscale simulations. This allows us to obtain unprecedented insight into relevant microscopic materials properties, in particular static and dynamic disorder contributions for a series of application-relevant molecules. We find that static disorder and thus the distribution of shallow traps is highly asymmetrical for many materials, impacting widely considered Gaussian disorder models. We furthermore analyse characteristic energy level fluctuation times and compare them to typical hopping rates to evaluate the importance of dynamic disorder for charge transport. We hope that our findings will significantly improve the accuracy of computational methods used to predict application relevant materials properties of organic semiconductors, and thus make these methods applicable for virtual materials design.