 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInterpretable Causal Representation Learning for Biological Data in the Pathway Space
Jun 14, 2025



Predicting the impact of genomic and drug perturbations in cellular function is crucial for understanding gene functions and drug effects, ultimately leading to improved therapies. To this end, Causal Representation Learning (CRL) constitutes one of the most promising approaches, as it aims to identify the latent factors that causally govern biological systems, thus facilitating the prediction of the effect of unseen perturbations. Yet, current CRL methods fail in reconciling their principled latent representations with known biological processes, leading to models that are not interpretable. To address this major issue, we present SENA-discrepancy-VAE, a model based on the recently proposed CRL method discrepancy-VAE, that produces representations where each latent factor can be interpreted as the (linear) combination of the activity of a (learned) set of biological processes. To this extent, we present an encoder, SENA-{\delta}, that efficiently compute and map biological processes' activity levels to the latent causal factors. We show that SENA-discrepancy-VAE achieves predictive performances on unseen combinations of interventions that are comparable with its original, non-interpretable counterpart, while inferring causal latent factors that are biologically meaningful.
Feature Selection with the R Package MXM: Discovering Statistically-Equivalent Feature Subsets
Nov 10, 2016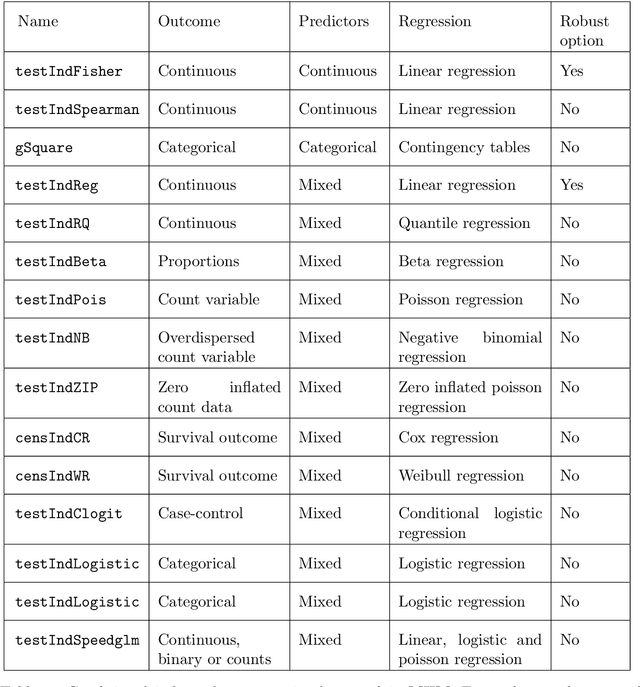
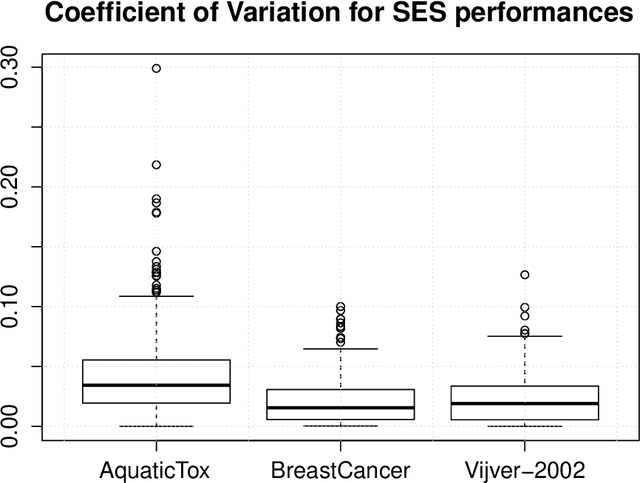
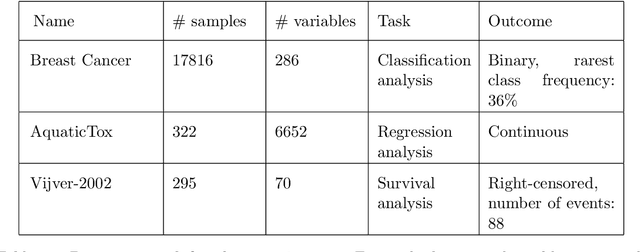
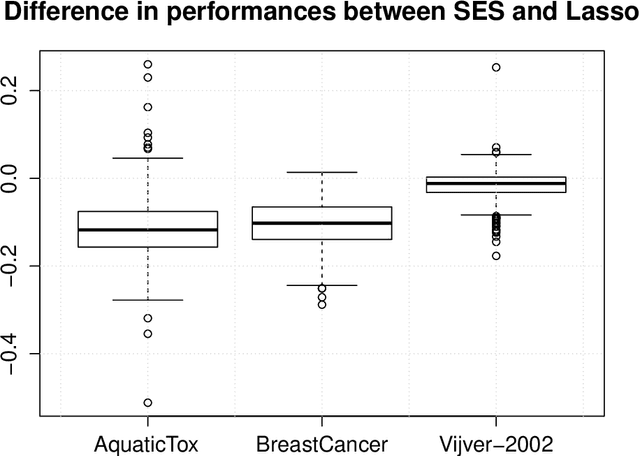
The statistically equivalent signature (SES) algorithm is a method for feature selection inspired by the principles of constrained-based learning of Bayesian Networks. Most of the currently available feature-selection methods return only a single subset of features, supposedly the one with the highest predictive power. We argue that in several domains multiple subsets can achieve close to maximal predictive accuracy, and that arbitrarily providing only one has several drawbacks. The SES method attempts to identify multiple, predictive feature subsets whose performances are statistically equivalent. Under that respect SES subsumes and extends previous feature selection algorithms, like the max-min parent children algorithm. SES is implemented in an homonym function included in the R package MXM, standing for mens ex machina, meaning 'mind from the machine' in Latin. The MXM implementation of SES handles several data-analysis tasks, namely classification, regression and survival analysis. In this paper we present the SES algorithm, its implementation, and provide examples of use of the SES function in R. Furthermore, we analyze three publicly available data sets to illustrate the equivalence of the signatures retrieved by SES and to contrast SES against the state-of-the-art feature selection method LASSO. Our results provide initial evidence that the two methods perform comparably well in terms of predictive accuracy and that multiple, equally predictive signatures are actually present in real world data.