 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA standard transformer and attention with linear biases for molecular conformer generation
Jun 24, 2025Sampling low-energy molecular conformations, spatial arrangements of atoms in a molecule, is a critical task for many different calculations performed in the drug discovery and optimization process. Numerous specialized equivariant networks have been designed to generate molecular conformations from 2D molecular graphs. Recently, non-equivariant transformer models have emerged as a viable alternative due to their capability to scale to improve generalization. However, the concern has been that non-equivariant models require a large model size to compensate the lack of equivariant bias. In this paper, we demonstrate that a well-chosen positional encoding effectively addresses these size limitations. A standard transformer model incorporating relative positional encoding for molecular graphs when scaled to 25 million parameters surpasses the current state-of-the-art non-equivariant base model with 64 million parameters on the GEOM-DRUGS benchmark. We implemented relative positional encoding as a negative attention bias that linearly increases with the shortest path distances between graph nodes at varying slopes for different attention heads, similar to ALiBi, a widely adopted relative positional encoding technique in the NLP domain. This architecture has the potential to serve as a foundation for a novel class of generative models for molecular conformations.
BMFM-RNA: An Open Framework for Building and Evaluating Transcriptomic Foundation Models
Jun 17, 2025



Transcriptomic foundation models (TFMs) have recently emerged as powerful tools for analyzing gene expression in cells and tissues, supporting key tasks such as cell-type annotation, batch correction, and perturbation prediction. However, the diversity of model implementations and training strategies across recent TFMs, though promising, makes it challenging to isolate the contribution of individual design choices or evaluate their potential synergies. This hinders the field's ability to converge on best practices and limits the reproducibility of insights across studies. We present BMFM-RNA, an open-source, modular software package that unifies diverse TFM pretraining and fine-tuning objectives within a single framework. Leveraging this capability, we introduce a novel training objective, whole cell expression decoder (WCED), which captures global expression patterns using an autoencoder-like CLS bottleneck representation. In this paper, we describe the framework, supported input representations, and training objectives. We evaluated four model checkpoints pretrained on CELLxGENE using combinations of masked language modeling (MLM), WCED and multitask learning. Using the benchmarking capabilities of BMFM-RNA, we show that WCED-based models achieve performance that matches or exceeds state-of-the-art approaches like scGPT across more than a dozen datasets in both zero-shot and fine-tuning tasks. BMFM-RNA, available as part of the biomed-multi-omics project ( https://github.com/BiomedSciAI/biomed-multi-omic ), offers a reproducible foundation for systematic benchmarking and community-driven exploration of optimal TFM training strategies, enabling the development of more effective tools to leverage the latest advances in AI for understanding cell biology.
Integration of AI and mechanistic modeling in generative adversarial networks for stochastic inverse problems
Sep 17, 2020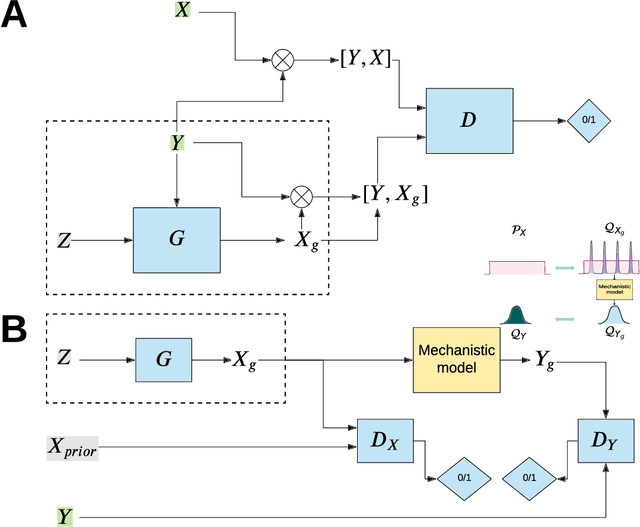
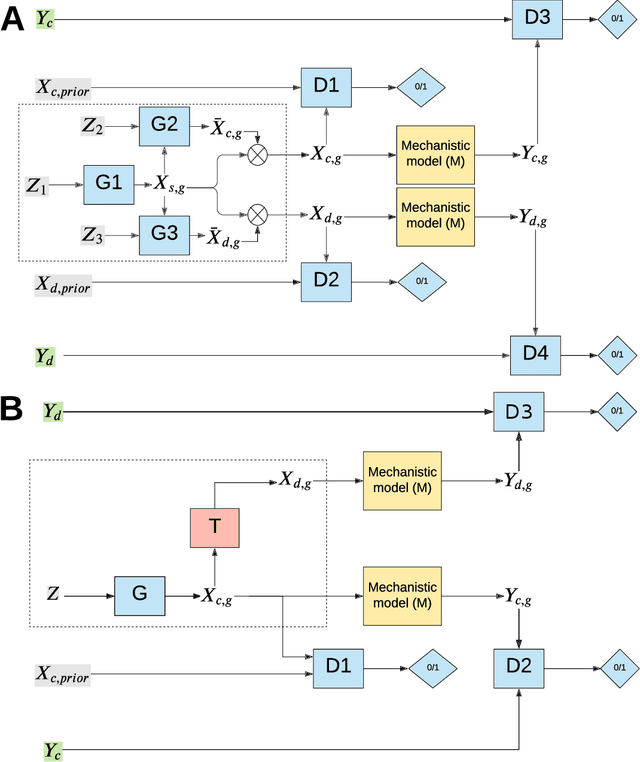
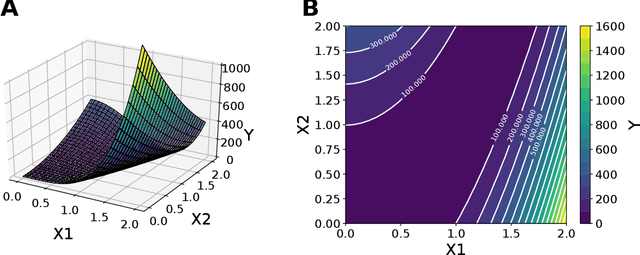
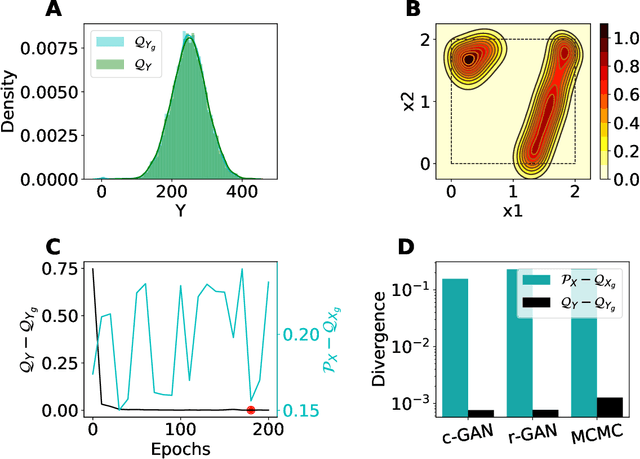
The problem of finding distributions of input parameters for deterministic mechanistic models to match distributions of model outputs to stochastic observations, i.e., the "Stochastic Inverse Problem" (SIP), encompasses a range of common tasks across a variety of scientific disciplines. Here, we demonstrate that SIP could be reformulated as a constrained optimization problem and adapted for applications in intervention studies to simultaneously infer model input parameters for two sets of observations, under control conditions and under an intervention. In the constrained optimization problem, the solution of SIP is enforced to accommodate the prior knowledge on the model input parameters and to produce outputs consistent with given observations by minimizing the divergence between the inferred distribution of input parameters and the prior. Unlike in standard SIP, the prior incorporates not only knowledge about model input parameters for objects in each set, but also information on the joint distribution or the deterministic map between the model input parameters in two sets of observations. To solve standard and intervention SIP, we employed conditional generative adversarial networks (GANs) and designed novel GANs that incorporate multiple generators and discriminators and have structures that reflect the underlying constrained optimization problems. This reformulation allows us to build computationally scalable solutions to tackle complex model input parameter inference scenarios, which appear routinely in physics, biophysics, economics and other areas, and which currently could not be handled with existing methods.
Beyond Backprop: Online Alternating Minimization with Auxiliary Variables
Oct 24, 2018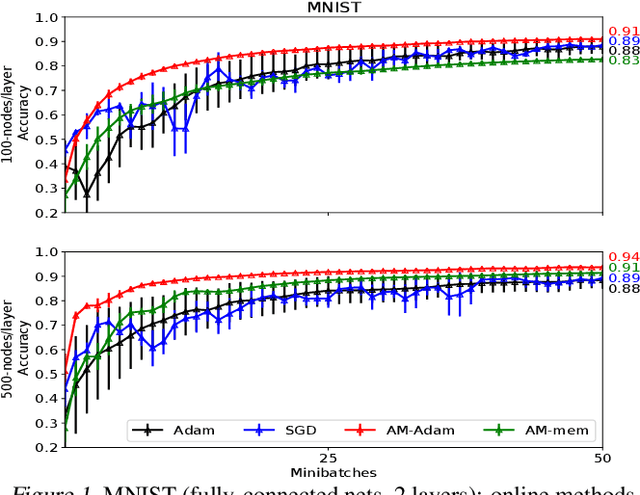
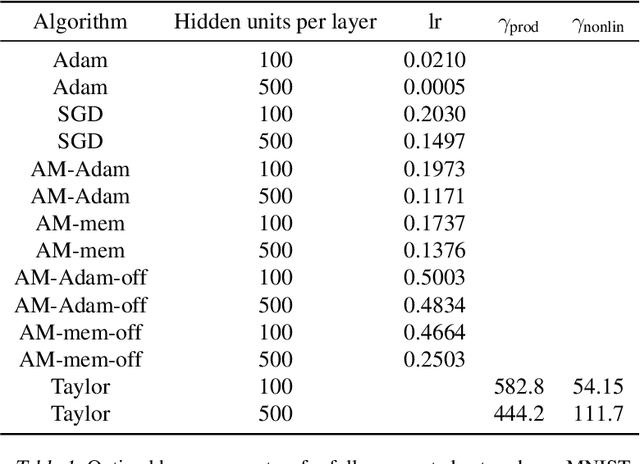
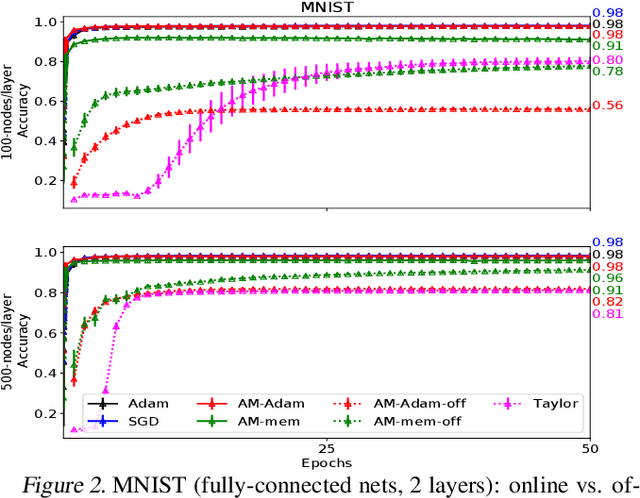
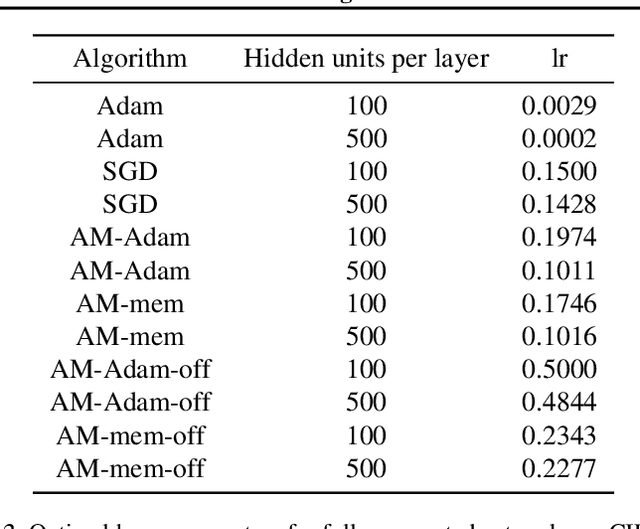
We propose a novel online alternating minimization (AltMin) algorithm for training deep neural networks, provide theoretical convergence guarantees and demonstrate its advantages on several classification tasks as compared both to standard backpropagation with stochastic gradient descent (backprop-SGD) and to offline alternating minimization. The key difference from backpropagation is an explicit optimization over hidden activations, which eliminates gradient chain computation in backprop, and breaks the weight training problem into independent, local optimization subproblems; this allows to avoid vanishing gradient issues, simplify handling non-differentiable nonlinearities, and perform parallel weight updates across the layers. Moreover, parallel local synaptic weight optimization with explicit activation propagation is a step closer to a more biologically plausible learning model than backpropagation, whose biological implausibility has been frequently criticized. Finally, the online nature of our approach allows to handle very large datasets, as well as continual, lifelong learning, which is our key contribution on top of recently proposed offline alternating minimization schemes (e.g., (Carreira-Perpinan andWang 2014), (Taylor et al. 2016)).