 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePerformance Analysis of Clustering Algorithms for Gene Expression Data
Jul 12, 2013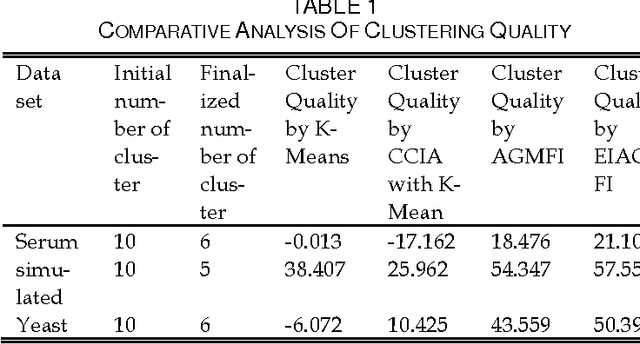
Microarray technology is a process that allows thousands of genes simultaneously monitor to various experimental conditions. It is used to identify the co-expressed genes in specific cells or tissues that are actively used to make proteins, This method is used to analysis the gene expression, an important task in bioinformatics research. Cluster analysis of gene expression data has proved to be a useful tool for identifying co-expressed genes, biologically relevant groupings of genes and samples. In this paper we analysed K-Means with Automatic Generations of Merge Factor for ISODATA- AGMFI, to group the microarray data sets on the basic of ISODATA. AGMFI is to generate initial values for merge and Spilt factor, maximum merge times instead of selecting efficient values as in ISODATA. The initial seeds for each cluster were normally chosen either sequentially or randomly. The quality of the final clusters was found to be influenced by these initial seeds. For the real life problems, the suitable number of clusters cannot be predicted. To overcome the above drawback the current research focused on developing the clustering algorithms without giving the initial number of clusters.
* 4 pages,4 figures. arXiv admin note: substantial text overlap with arXiv:1112.4261, arXiv:1201.4914, arXiv:1307.3337
Unsupervised Gene Expression Data using Enhanced Clustering Method
Jul 12, 2013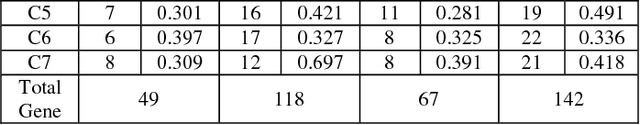
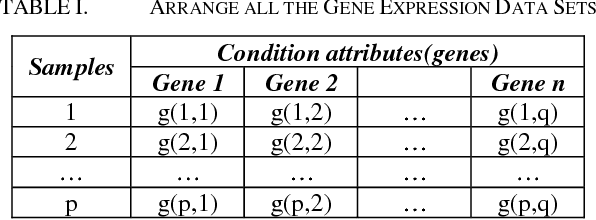
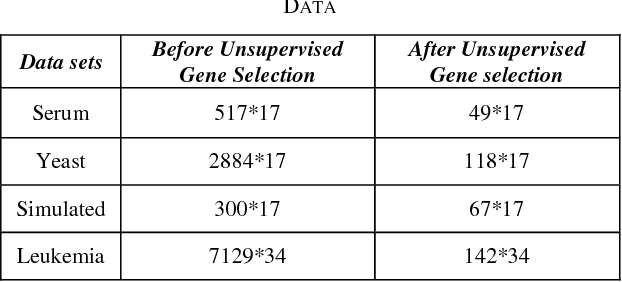
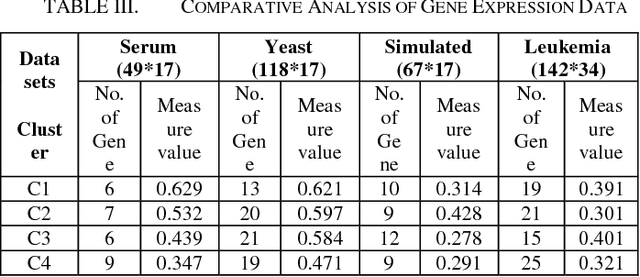
Microarrays are made it possible to simultaneously monitor the expression profiles of thousands of genes under various experimental conditions. Identification of co-expressed genes and coherent patterns is the central goal in microarray or gene expression data analysis and is an important task in bioinformatics research. Feature selection is a process to select features which are more informative. It is one of the important steps in knowledge discovery. The problem is that not all features are important. Some of the features may be redundant, and others may be irrelevant and noisy. In this work the unsupervised Gene selection method and Enhanced Center Initialization Algorithm (ECIA) with K-Means algorithms have been applied for clustering of Gene Expression Data. This proposed clustering algorithm overcomes the drawbacks in terms of specifying the optimal number of clusters and initialization of good cluster centroids. Gene Expression Data show that could identify compact clusters with performs well in terms of the Silhouette Coefficients cluster measure.
* 5 pages, 1 figures, conference
A Novel Approach for Single Gene Selection Using Clustering and Dimensionality Reduction
Jun 10, 2013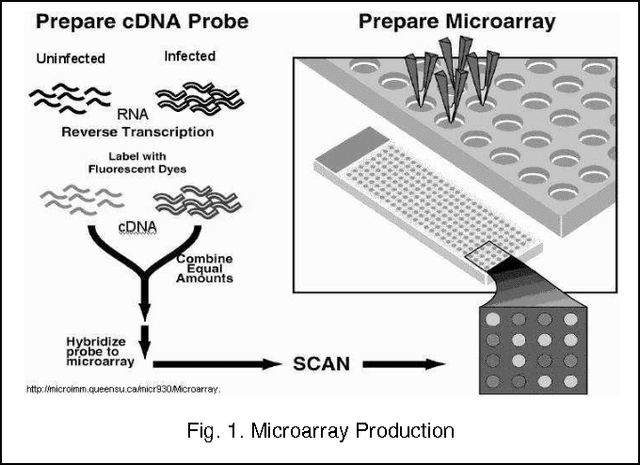

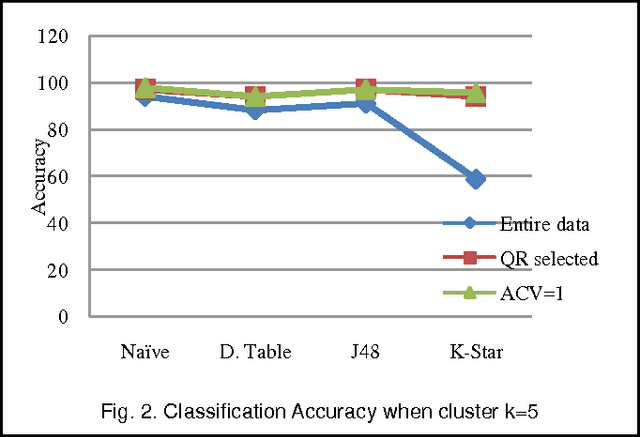
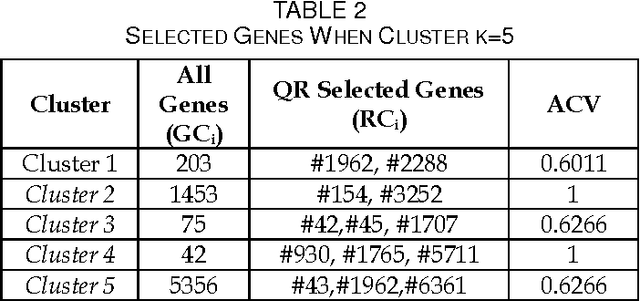
We extend the standard rough set-based approach to deal with huge amounts of numeric attributes versus small amount of available objects. Here, a novel approach of clustering along with dimensionality reduction; Hybrid Fuzzy C Means-Quick Reduct (FCMQR) algorithm is proposed for single gene selection. Gene selection is a process to select genes which are more informative. It is one of the important steps in knowledge discovery. The problem is that all genes are not important in gene expression data. Some of the genes may be redundant, and others may be irrelevant and noisy. In this study, the entire dataset is divided in proper grouping of similar genes by applying Fuzzy C Means (FCM) algorithm. A high class discriminated genes has been selected based on their degree of dependence by applying Quick Reduct algorithm based on Rough Set Theory to all the resultant clusters. Average Correlation Value (ACV) is calculated for the high class discriminated genes. The clusters which have the ACV value a s 1 is determined as significant clusters, whose classification accuracy will be equal or high when comparing to the accuracy of the entire dataset. The proposed algorithm is evaluated using WEKA classifiers and compared. Finally, experimental results related to the leukemia cancer data confirm that our approach is quite promising, though it surely requires further research.
* 6 pages, 4 figures. arXiv admin note: text overlap with arXiv:1306.1323
Verdict Accuracy of Quick Reduct Algorithm using Clustering and Classification Techniques for Gene Expression Data
Jun 06, 2013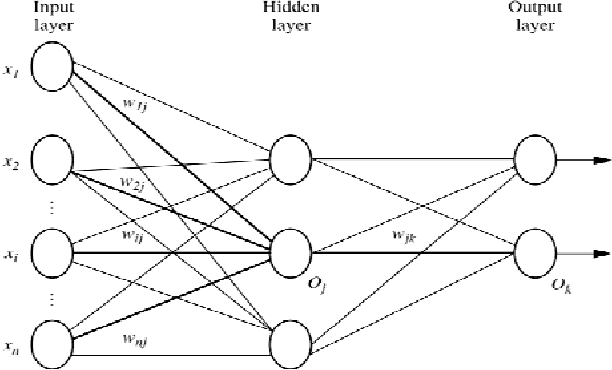

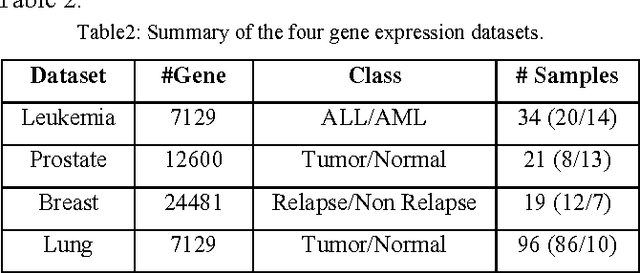
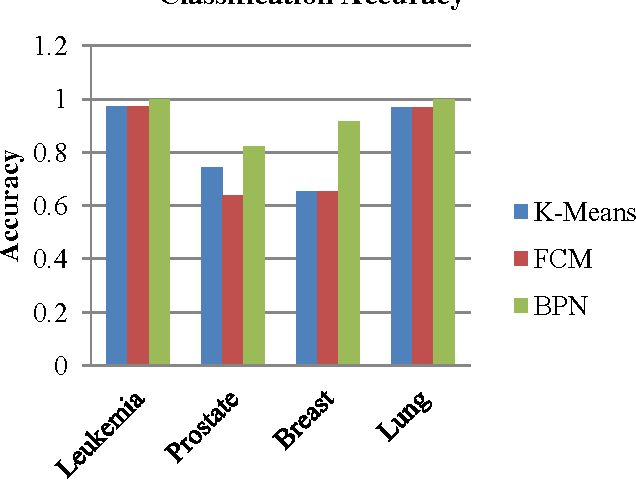
In most gene expression data, the number of training samples is very small compared to the large number of genes involved in the experiments. However, among the large amount of genes, only a small fraction is effective for performing a certain task. Furthermore, a small subset of genes is desirable in developing gene expression based diagnostic tools for delivering reliable and understandable results. With the gene selection results, the cost of biological experiment and decision can be greatly reduced by analyzing only the marker genes. An important application of gene expression data in functional genomics is to classify samples according to their gene expression profiles. Feature selection (FS) is a process which attempts to select more informative features. It is one of the important steps in knowledge discovery. Conventional supervised FS methods evaluate various feature subsets using an evaluation function or metric to select only those features which are related to the decision classes of the data under consideration. This paper studies a feature selection method based on rough set theory. Further K-Means, Fuzzy C-Means (FCM) algorithm have implemented for the reduced feature set without considering class labels. Then the obtained results are compared with the original class labels. Back Propagation Network (BPN) has also been used for classification. Then the performance of K-Means, FCM, and BPN are analyzed through the confusion matrix. It is found that the BPN is performing well comparatively.
* 7 pages, 3 figures
Performance Analysis of Enhanced Clustering Algorithm for Gene Expression Data
Dec 19, 2011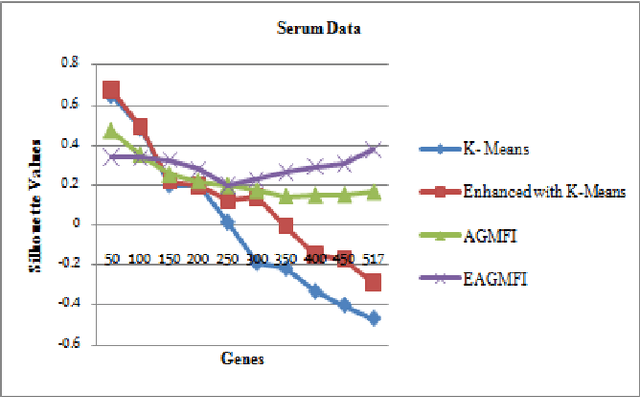
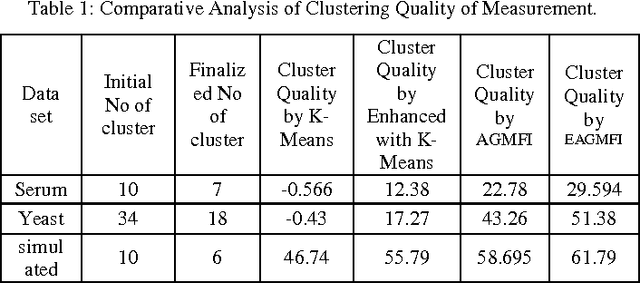
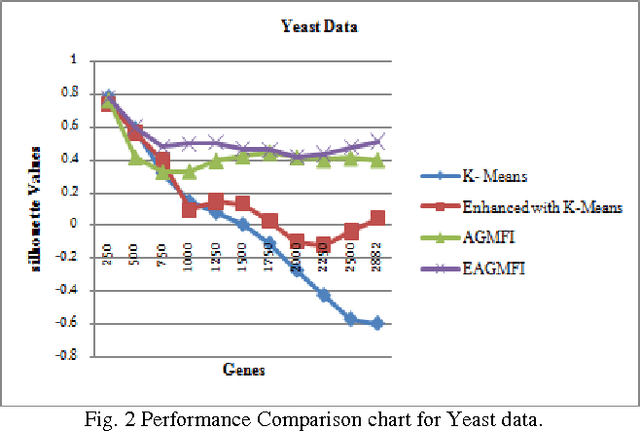
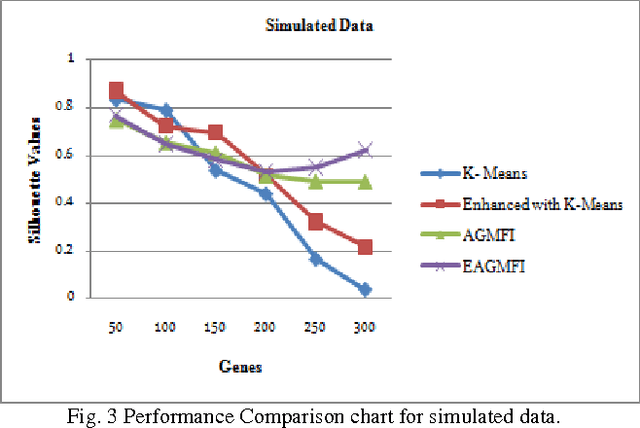
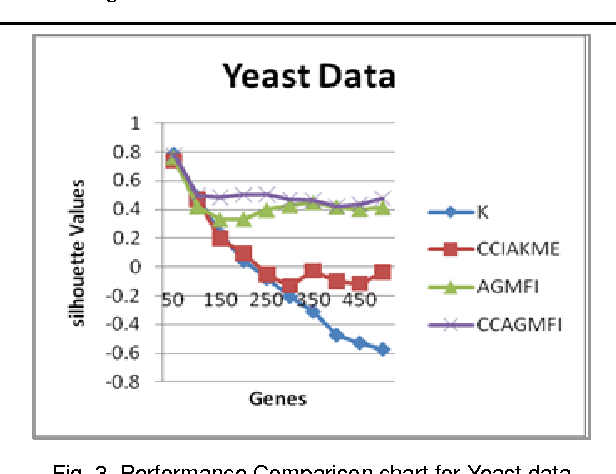
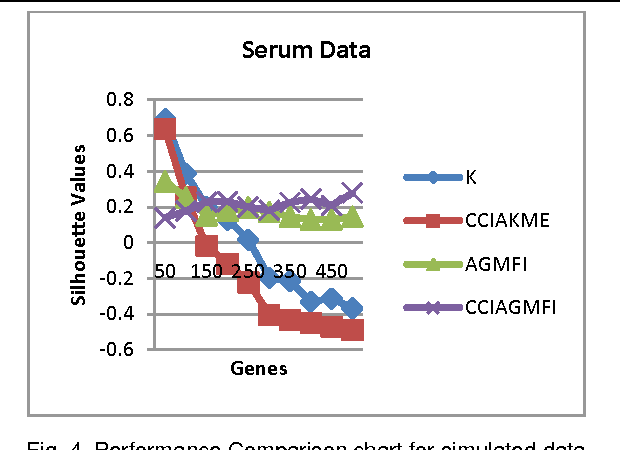
Microarrays are made it possible to simultaneously monitor the expression profiles of thousands of genes under various experimental conditions. It is used to identify the co-expressed genes in specific cells or tissues that are actively used to make proteins. This method is used to analysis the gene expression, an important task in bioinformatics research. Cluster analysis of gene expression data has proved to be a useful tool for identifying co-expressed genes, biologically relevant groupings of genes and samples. In this paper we applied K-Means with Automatic Generations of Merge Factor for ISODATA- AGMFI. Though AGMFI has been applied for clustering of Gene Expression Data, this proposed Enhanced Automatic Generations of Merge Factor for ISODATA- EAGMFI Algorithms overcome the drawbacks of AGMFI in terms of specifying the optimal number of clusters and initialization of good cluster centroids. Experimental results on Gene Expression Data show that the proposed EAGMFI algorithms could identify compact clusters with perform well in terms of the Silhouette Coefficients cluster measure.
* ISSN (Online): 1694-0814 http://www.IJCSI.org