 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMass Classification Method in Mammogram Using Fuzzy K-Nearest Neighbour Equality
Jun 18, 2014
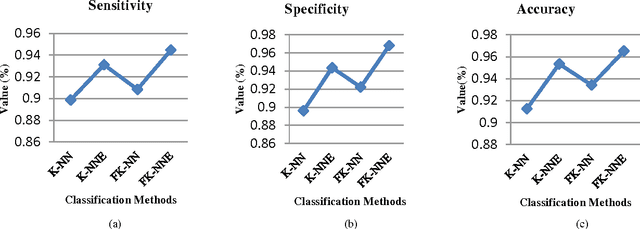

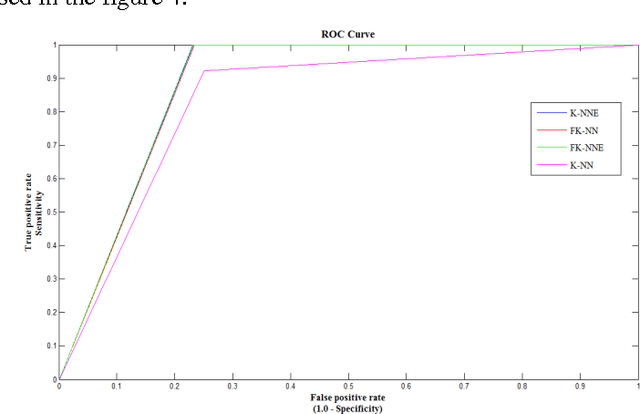
Mass classification of objects is an important area of research and application in a variety of fields. In this paper, we present an efficient computer aided mass classification method in digitized mammograms using Fuzzy K-Nearest Neighbor Equality, which performs benign or malignant classification on region of interest that contains mass. One of the major mammographic characteristics for mass classification is texture. Fuzzy K-Nearest Neighbor Equality exploits this important factor to classify the mass into benign or malignant. The statistical textural features used in characterizing the masses are Haralick and Run length features. The main aim of the method is to increase the effectiveness and efficiency of the classification process in an objective manner to reduce the numbers of false positive of malignancies. In this paper proposes a novel Fuzzy K-Nearest Neighbor Equality algorithm for classifying the marked regions into benign and malignant and 94.46 sensitivity,96.81 specificity and 96.52 accuracy is achieved that is very much promising compare to the radiologists' accuracy.
Pectoral Muscles Suppression in Digital Mammograms using Hybridization of Soft Computing Methods
Jan 05, 2014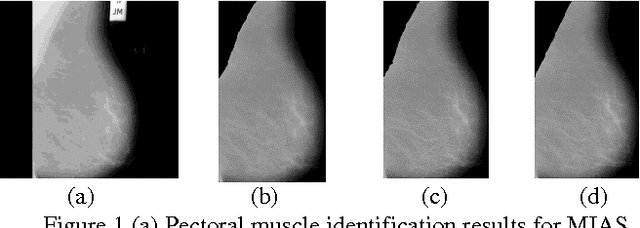
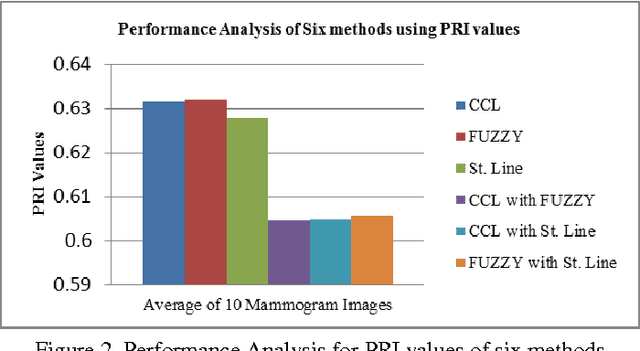
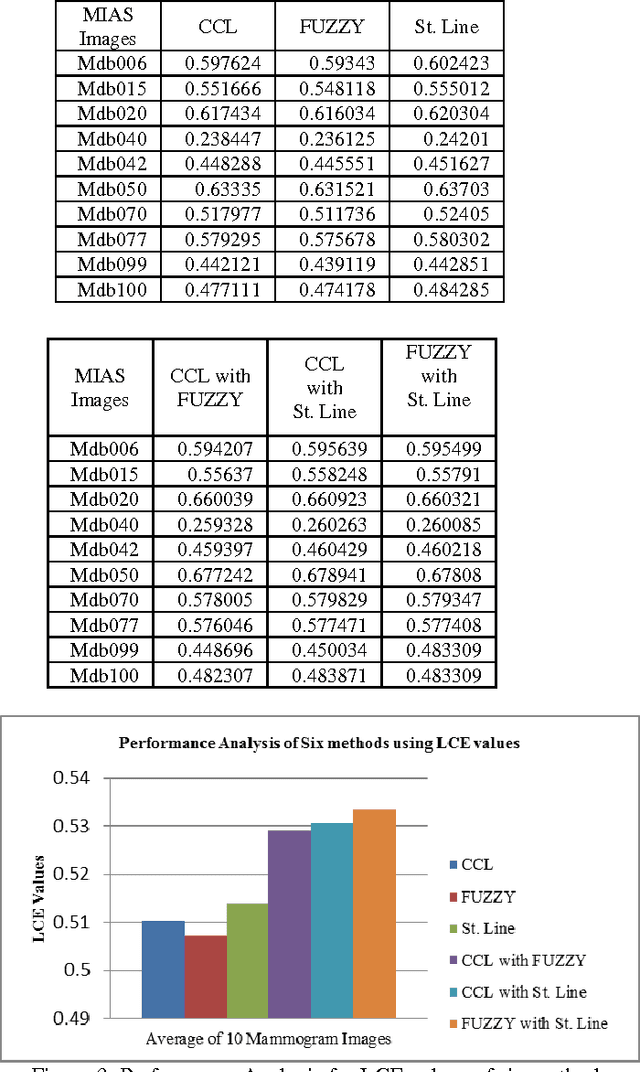
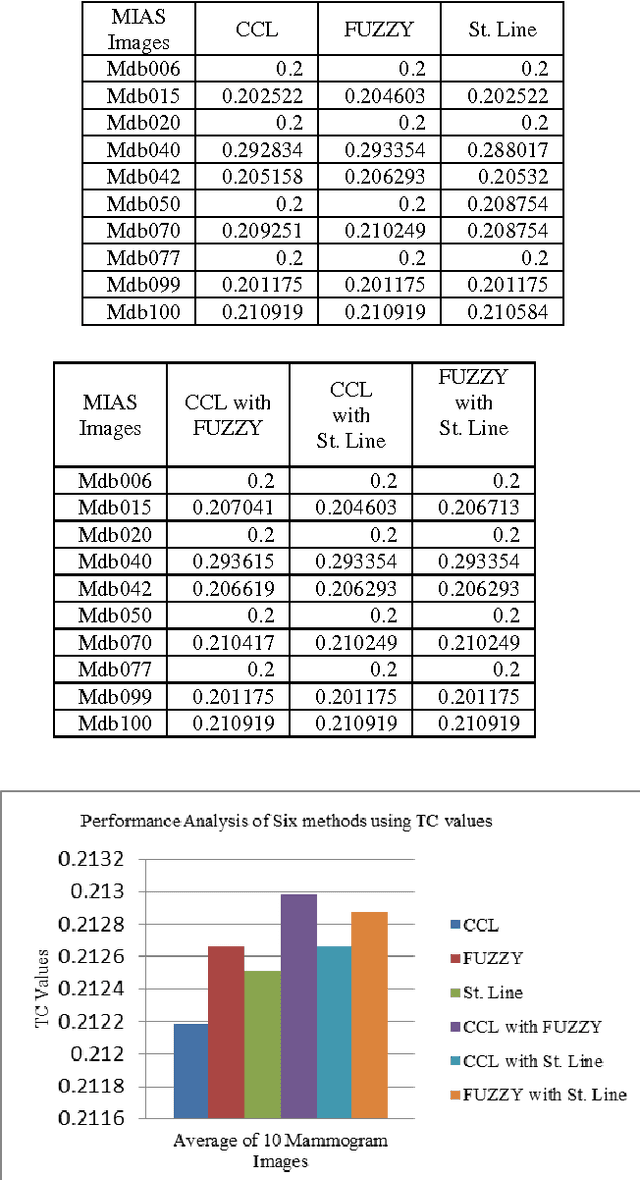
Breast region segmentation is an essential prerequisite in computerized analysis of mammograms. It aims at separating the breast tissue from the background of the mammogram and it includes two independent segmentations. The first segments the background region which usually contains annotations, labels and frames from the whole breast region, while the second removes the pectoral muscle portion (present in Medio Lateral Oblique (MLO) views) from the rest of the breast tissue. In this paper we propose hybridization of Connected Component Labeling (CCL), Fuzzy, and Straight line methods. Our proposed methods worked good for separating pectoral region. After removal pectoral muscle from the mammogram, further processing is confined to the breast region alone. To demonstrate the validity of our segmentation algorithm, it is extensively tested using over 322 mammographic images from the Mammographic Image Analysis Society (MIAS) database. The segmentation results were evaluated using a Mean Absolute Error (MAE), Hausdroff Distance (HD), Probabilistic Rand Index (PRI), Local Consistency Error (LCE) and Tanimoto Coefficient (TC). The hybridization of fuzzy with straight line method is given more than 96% of the curve segmentations to be adequate or better. In addition a comparison with similar approaches from the state of the art has been given, obtaining slightly improved results. Experimental results demonstrate the effectiveness of the proposed approach.
Automatic Mammogram image Breast Region Extraction and Removal of Pectoral Muscle
Jul 29, 2013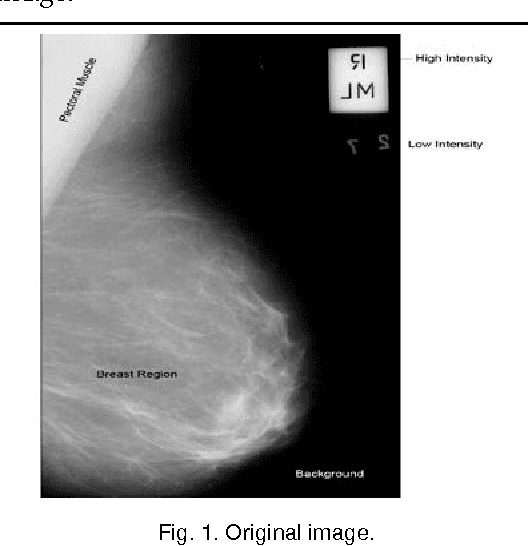
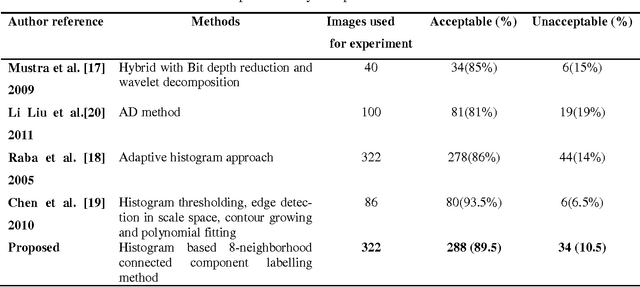
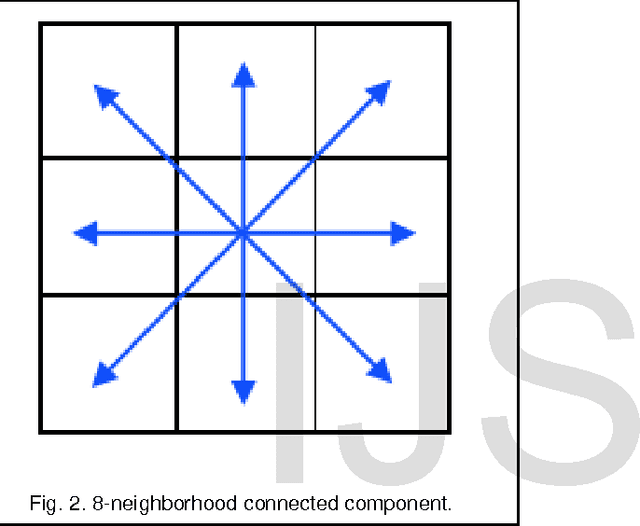
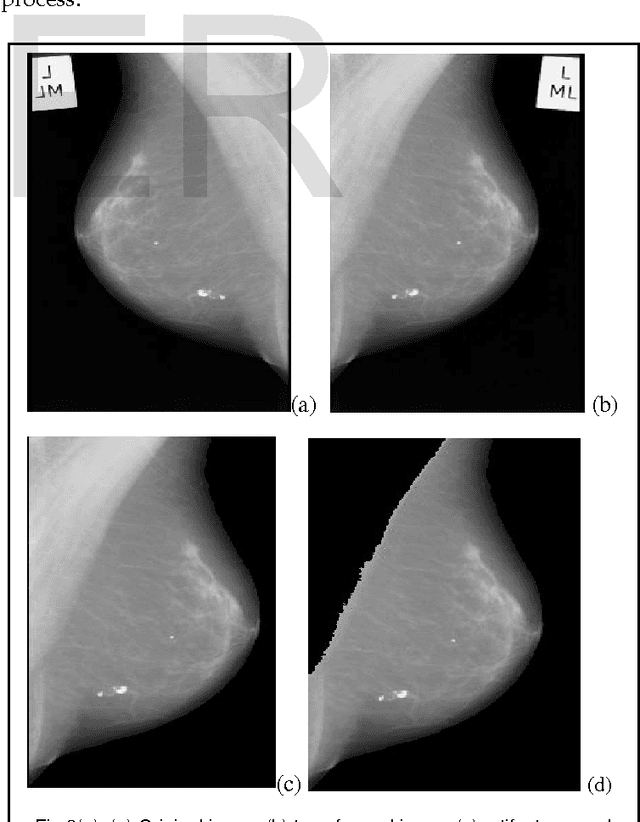
Currently Mammography is a most effective imaging modality used by radiologists for the screening of breast cancer. Finding an accurate, robust and efficient breast region segmentation technique still remains a challenging problem in digital mammography. Extraction of the breast profile region and the removal of pectoral muscle are essential pre-processing steps in Computer Aided Diagnosis (CAD) system for the diagnosis of breast cancer. Primarily it allows the search for abnormalities to be limited to the region of the breast tissue without undue influence from the background of the mammogram. The presence of pectoral muscle in mammograms biases detection procedures, which recommends removing the pectoral muscle during mammogram image pre-processing. The presence of pectoral muscle in mammograms may disturb or influence the detection of breast cancer as the pectoral muscle and mammographic parenchymas appear similar. The goal of breast region extraction is reducing the image size without losing anatomic information, it improve the accuracy of the overall CAD system. The main objective of this study is to propose an automated method to identify the pectoral muscle in Medio-Lateral Oblique (MLO) view mammograms. In this paper, we proposed histogram based 8-neighborhood connected component labelling method for breast region extraction and removal of pectoral muscle. The proposed method is evaluated by using the mean values of accuracy and error. The comparative analysis shows that the proposed method identifies the breast region more accurately.
* 8 Pages, 5 Figures
Mammogram Edge Detection Using Hybrid Soft Computing Methods
Jul 17, 2013

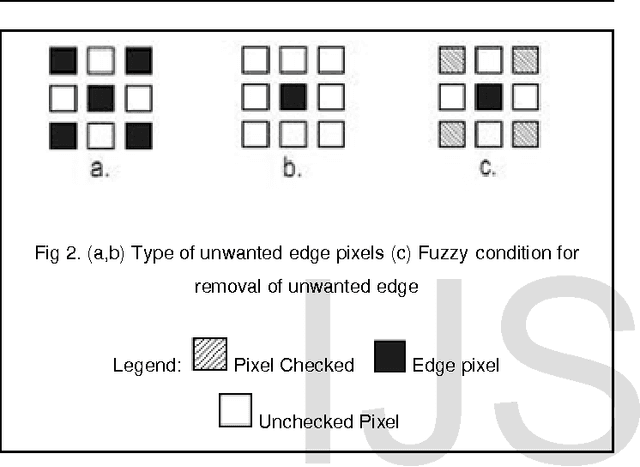
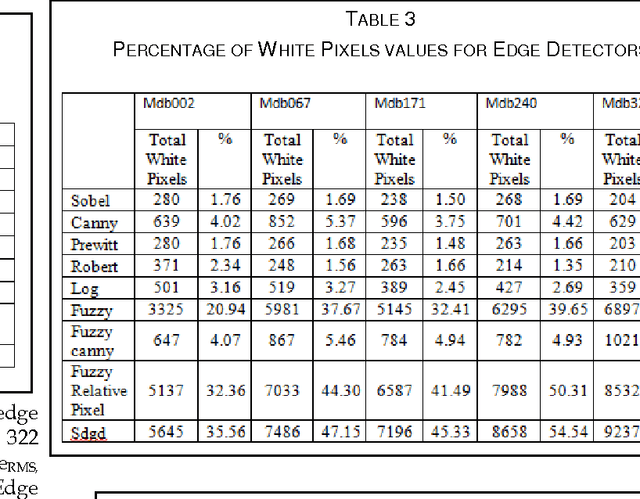
Image segmentation is a crucial step in a wide range of method image processing systems. It is useful in visualization of the different objects present in the image. In spite of the several methods available in the literature, image segmentation still a challenging problem in most of image processing applications. The challenge comes from the fuzziness of image objects and the overlapping of the different regions. Detection of edges in an image is a very important step towards understanding image features. There are large numbers of edge detection operators available, each designed to be sensitive to certain types of edges. The Quality of edge detection can be measured from several criteria objectively. Some criteria are proposed in terms of mathematical measurement, some of them are based on application and implementation requirements. Since edges often occur at image locations representing object boundaries, edge detection is extensively used in image segmentation when images are divided into areas corresponding to different objects. This can be used specifically for enhancing the tumor area in mammographic images. Different methods are available for edge detection like Roberts, Sobel, Prewitt, Canny, Log edge operators. In this paper a novel algorithms for edge detection has been proposed for mammographic images. Breast boundary, pectoral region and tumor location can be seen clearly by using this method. For comparison purpose Roberts, Sobel, Prewitt, Canny, Log edge operators are used and their results are displayed. Experimental results demonstrate the effectiveness of the proposed approach.
Performance Analysis of Clustering Algorithms for Gene Expression Data
Jul 12, 2013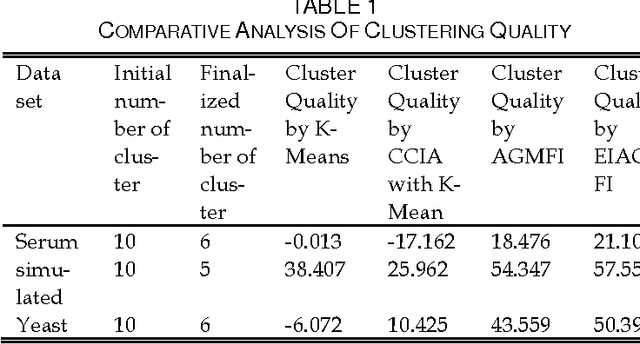
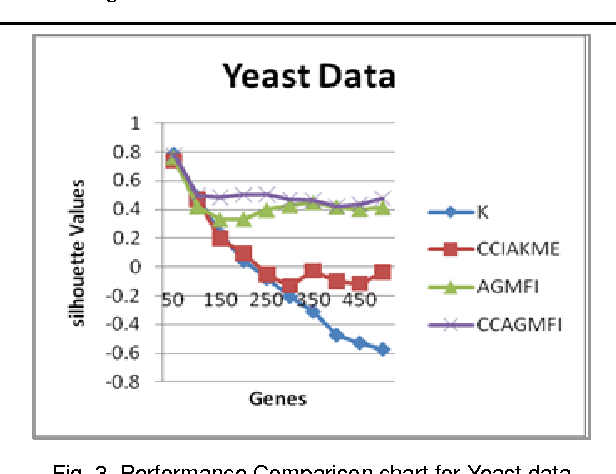
Microarray technology is a process that allows thousands of genes simultaneously monitor to various experimental conditions. It is used to identify the co-expressed genes in specific cells or tissues that are actively used to make proteins, This method is used to analysis the gene expression, an important task in bioinformatics research. Cluster analysis of gene expression data has proved to be a useful tool for identifying co-expressed genes, biologically relevant groupings of genes and samples. In this paper we analysed K-Means with Automatic Generations of Merge Factor for ISODATA- AGMFI, to group the microarray data sets on the basic of ISODATA. AGMFI is to generate initial values for merge and Spilt factor, maximum merge times instead of selecting efficient values as in ISODATA. The initial seeds for each cluster were normally chosen either sequentially or randomly. The quality of the final clusters was found to be influenced by these initial seeds. For the real life problems, the suitable number of clusters cannot be predicted. To overcome the above drawback the current research focused on developing the clustering algorithms without giving the initial number of clusters.
* 4 pages,4 figures. arXiv admin note: substantial text overlap with arXiv:1112.4261, arXiv:1201.4914, arXiv:1307.3337
Unsupervised Gene Expression Data using Enhanced Clustering Method
Jul 12, 2013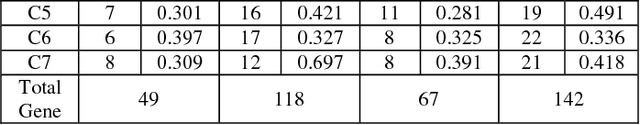
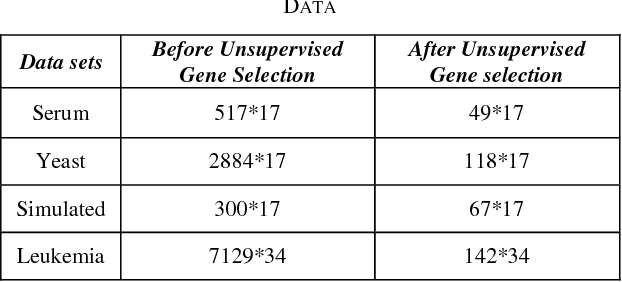
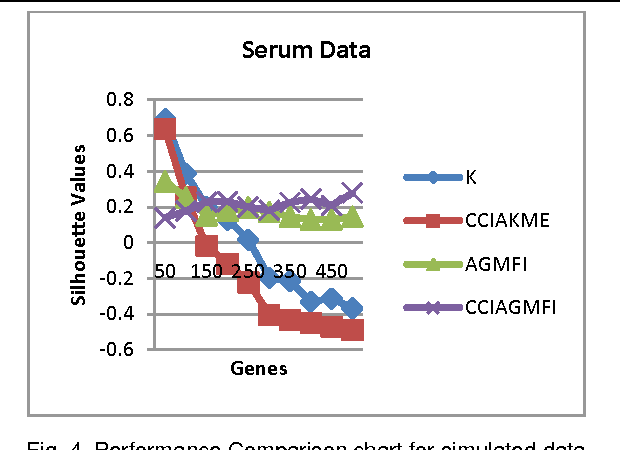
Microarrays are made it possible to simultaneously monitor the expression profiles of thousands of genes under various experimental conditions. Identification of co-expressed genes and coherent patterns is the central goal in microarray or gene expression data analysis and is an important task in bioinformatics research. Feature selection is a process to select features which are more informative. It is one of the important steps in knowledge discovery. The problem is that not all features are important. Some of the features may be redundant, and others may be irrelevant and noisy. In this work the unsupervised Gene selection method and Enhanced Center Initialization Algorithm (ECIA) with K-Means algorithms have been applied for clustering of Gene Expression Data. This proposed clustering algorithm overcomes the drawbacks in terms of specifying the optimal number of clusters and initialization of good cluster centroids. Gene Expression Data show that could identify compact clusters with performs well in terms of the Silhouette Coefficients cluster measure.
* 5 pages, 1 figures, conference
A Novel Approach for Single Gene Selection Using Clustering and Dimensionality Reduction
Jun 10, 2013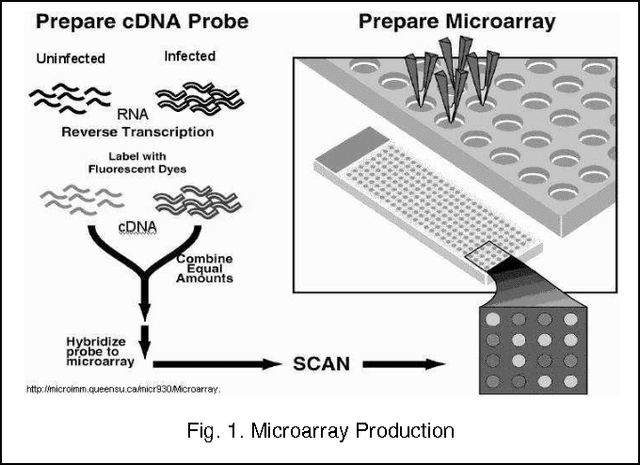

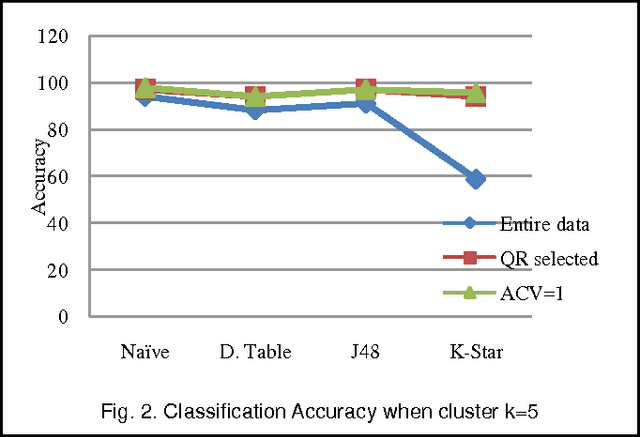
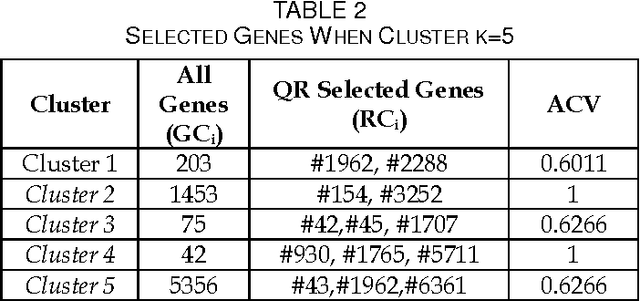
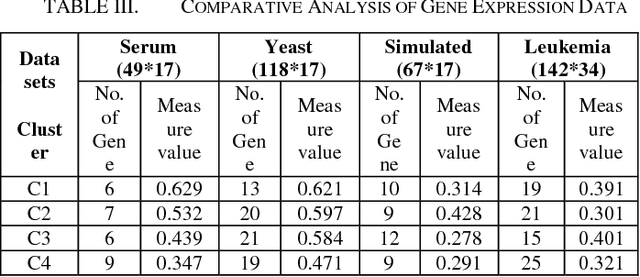
We extend the standard rough set-based approach to deal with huge amounts of numeric attributes versus small amount of available objects. Here, a novel approach of clustering along with dimensionality reduction; Hybrid Fuzzy C Means-Quick Reduct (FCMQR) algorithm is proposed for single gene selection. Gene selection is a process to select genes which are more informative. It is one of the important steps in knowledge discovery. The problem is that all genes are not important in gene expression data. Some of the genes may be redundant, and others may be irrelevant and noisy. In this study, the entire dataset is divided in proper grouping of similar genes by applying Fuzzy C Means (FCM) algorithm. A high class discriminated genes has been selected based on their degree of dependence by applying Quick Reduct algorithm based on Rough Set Theory to all the resultant clusters. Average Correlation Value (ACV) is calculated for the high class discriminated genes. The clusters which have the ACV value a s 1 is determined as significant clusters, whose classification accuracy will be equal or high when comparing to the accuracy of the entire dataset. The proposed algorithm is evaluated using WEKA classifiers and compared. Finally, experimental results related to the leukemia cancer data confirm that our approach is quite promising, though it surely requires further research.
* 6 pages, 4 figures. arXiv admin note: text overlap with arXiv:1306.1323
Verdict Accuracy of Quick Reduct Algorithm using Clustering and Classification Techniques for Gene Expression Data
Jun 06, 2013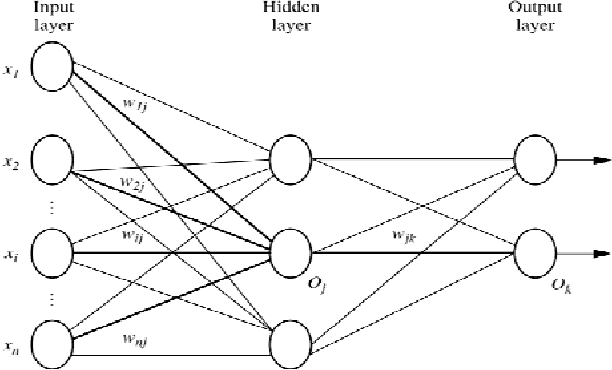

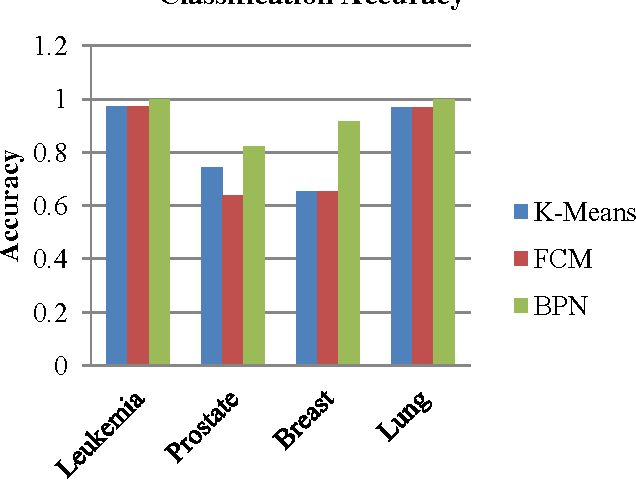
In most gene expression data, the number of training samples is very small compared to the large number of genes involved in the experiments. However, among the large amount of genes, only a small fraction is effective for performing a certain task. Furthermore, a small subset of genes is desirable in developing gene expression based diagnostic tools for delivering reliable and understandable results. With the gene selection results, the cost of biological experiment and decision can be greatly reduced by analyzing only the marker genes. An important application of gene expression data in functional genomics is to classify samples according to their gene expression profiles. Feature selection (FS) is a process which attempts to select more informative features. It is one of the important steps in knowledge discovery. Conventional supervised FS methods evaluate various feature subsets using an evaluation function or metric to select only those features which are related to the decision classes of the data under consideration. This paper studies a feature selection method based on rough set theory. Further K-Means, Fuzzy C-Means (FCM) algorithm have implemented for the reduced feature set without considering class labels. Then the obtained results are compared with the original class labels. Back Propagation Network (BPN) has also been used for classification. Then the performance of K-Means, FCM, and BPN are analyzed through the confusion matrix. It is found that the BPN is performing well comparatively.
* 7 pages, 3 figures
Fuzzy - Rough Feature Selection With Π- Membership Function For Mammogram Classification
May 24, 2012Breast cancer is the second leading cause for death among women and it is diagnosed with the help of mammograms. Oncologists are miserably failed in identifying the micro calcification at the early stage with the help of the mammogram visually. In order to improve the performance of the breast cancer screening, most of the researchers have proposed Computer Aided Diagnosis using image processing. In this study mammograms are preprocessed and features are extracted, then the abnormality is identified through the classification. If all the extracted features are used, most of the cases are misidentified. Hence feature selection procedure is sought. In this paper, Fuzzy-Rough feature selection with {\pi} membership function is proposed. The selected features are used to classify the abnormalities with help of Ant-Miner and Weka tools. The experimental analysis shows that the proposed method improves the mammograms classification accuracy.
Performance Analysis of Enhanced Clustering Algorithm for Gene Expression Data
Dec 19, 2011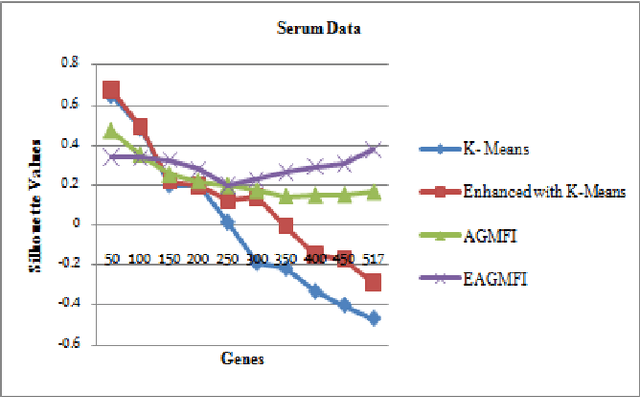
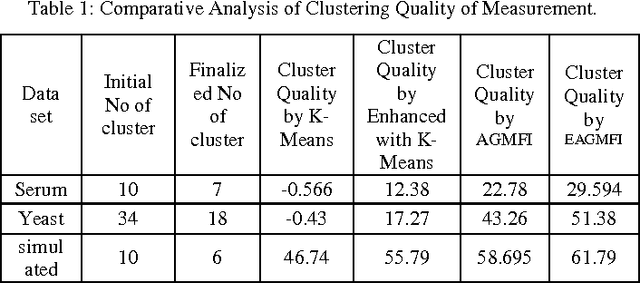
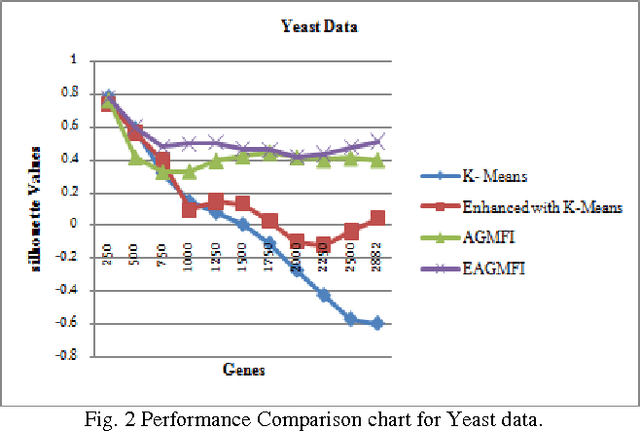
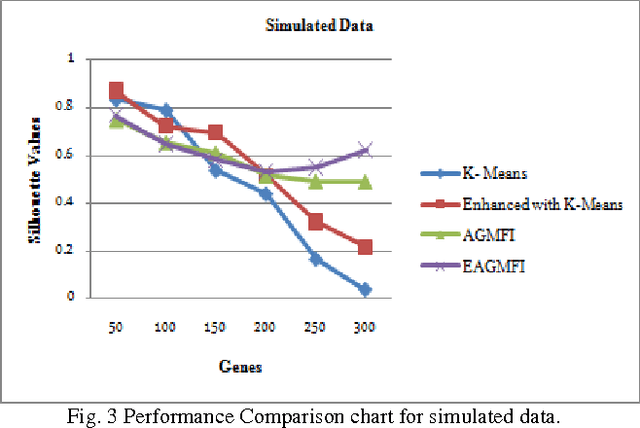
Microarrays are made it possible to simultaneously monitor the expression profiles of thousands of genes under various experimental conditions. It is used to identify the co-expressed genes in specific cells or tissues that are actively used to make proteins. This method is used to analysis the gene expression, an important task in bioinformatics research. Cluster analysis of gene expression data has proved to be a useful tool for identifying co-expressed genes, biologically relevant groupings of genes and samples. In this paper we applied K-Means with Automatic Generations of Merge Factor for ISODATA- AGMFI. Though AGMFI has been applied for clustering of Gene Expression Data, this proposed Enhanced Automatic Generations of Merge Factor for ISODATA- EAGMFI Algorithms overcome the drawbacks of AGMFI in terms of specifying the optimal number of clusters and initialization of good cluster centroids. Experimental results on Gene Expression Data show that the proposed EAGMFI algorithms could identify compact clusters with perform well in terms of the Silhouette Coefficients cluster measure.
* ISSN (Online): 1694-0814 http://www.IJCSI.org